奥利司他结构与光谱性质的密度泛函理论研究
2020-09-09李荫徐裕深梁小蕊
李荫 徐裕深 梁小蕊


摘 要:采用密度泛函理论B3LYP方法,研究了奥利司他分子。用 6-31G(d,p)基组计算了奥利司他分子的最稳定构型。在最稳定构型的基础上采用同样的理论方法计算得到了奥利司他分子的红外振动频率、核磁共振氢谱(1H NMR)和碳谱(13C NMR)。根据红外吸收峰强度将红外光谱分成5个区域,讨论了各区域的振动模式,并讨论了奥利司他分子的核磁共振1HNMR和13CNMR化学位移数据。
关 键 词:奥利司他;密度泛函理论;红外光谱;核磁共振
中图分类号:O657.3 文献标识码: A 文章编号: 1671-0460(2020)08-1630-05
Abstract: The molecule of orlistat was studied by density functional theory method. The stable structure and molecular orbital of orlistat were calculated by the method of B3LYP at 6-31G(d,p) level. Based on the stable configuration, the infrared vibration frequency, 13C NMR and 1H NMR of orlistat molecule were calculated by the same method. According to the vibration intensity, the infrared spectroscopy (IR) were divided into five regions, and their vibration mode were studied respectively. Then chemical shift of 13C NMR and 1H NMR were analyzed.
Key words: Orlistat; Density functional theory; Infrared spectra; 1H NMR spectra; 13C NMR spectra
奧利司他(Orlistat)为非作用于中枢神经系统的肥胖症治疗药,是美国FDA于1999年批准,用于肥胖症长期治疗的新药药物[1-2]。该药仅作用于胃肠道,通过抑制胃肠道的脂肪酶(脂肪酶为胃肠道分解脂肪所必需的酶,本品可与胃、胰脂肪酶的丝氨酸残基结合,使脂肪酶失活,使其不能将食物中的脂肪分解为游离脂肪酸,抑制脂肪的利用和吸收),阻止三酰甘油水解为游离脂肪酸和单酰基甘油酯,减少肠腔黏膜对膳食中脂肪(三酰甘油)的吸收,促使脂肪排出体外[3-9]。此外,本品尚可降低与肥胖症相关的危险因素。
近几年,我国食品市场上出现的减肥类保健食品日益增多,许多不法厂家为获得明显的疗效,在保健食品中添加此类处方药成分以达到速效、强效的目的。因此,为保护消费者的健康,对此类保健食品进行定性和定量检测,成为保障减肥药品市场安全的有效方法[10-11]。
1 计算方法
本文基于Gaussian09程序,采用密度泛函理论中的杂化密度泛函B3LYP方法,选取6-31G(d,p)基组,优化得到了奥利司他分子的稳定构型。在最稳定构型的基础上采用同样的理论方法计算得到了奥利司他分子的红外振动频率、核磁共振氢谱、碳谱,并进一步探讨了奥利司他分子的光谱特征。
2 结果与讨论
2.1 分子的几何构型
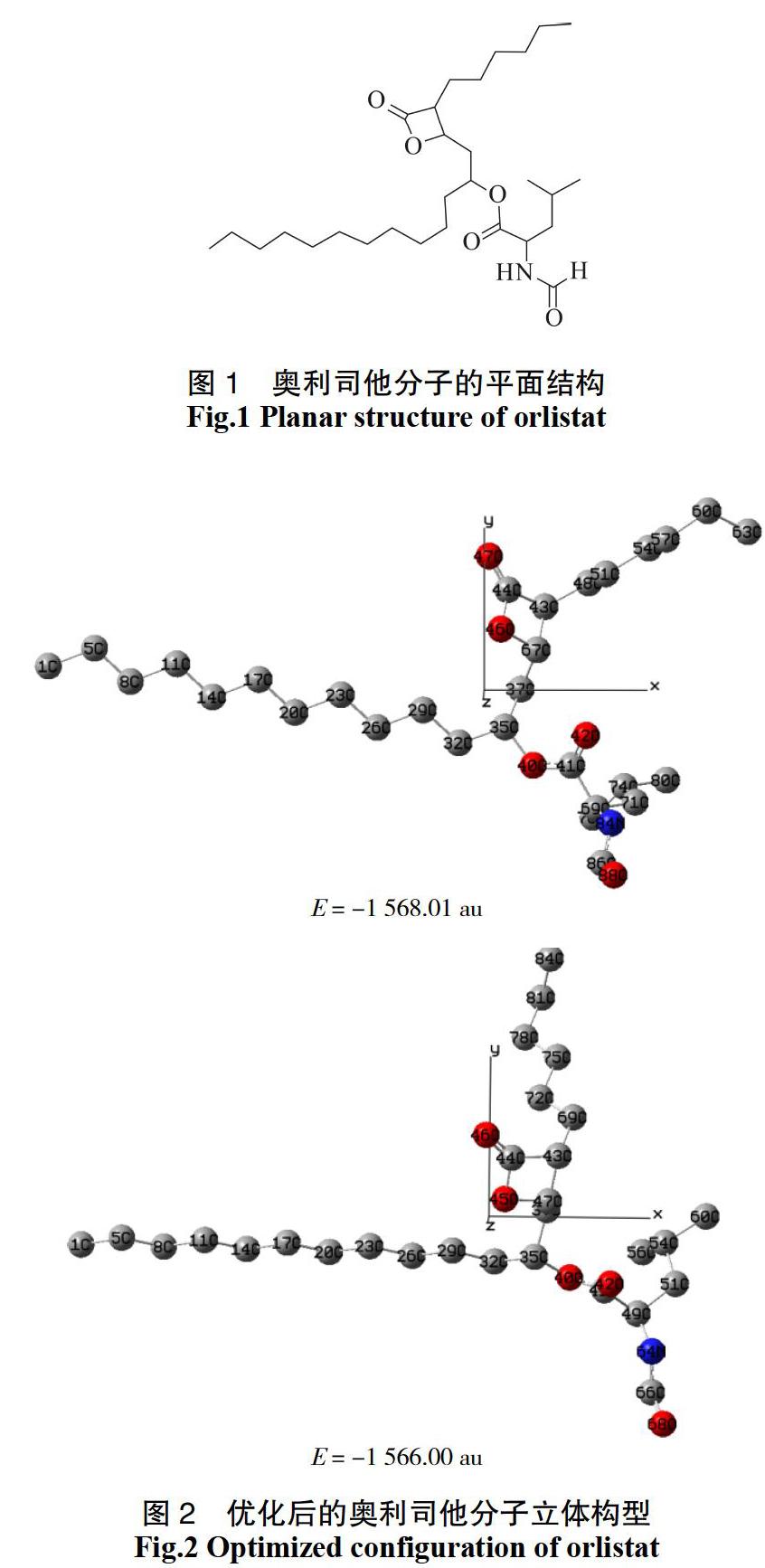
图1是奥利司他分子的平面结构,采用密度泛函理论方法B3LYP/6-31G(d,p)基组对奥利司他分子进行了几何结构全优化,优化后得到两种稳定构型,其原子编号及笛卡尔坐标系见图2。计算结果显示这两种构型的最低能量分别为E =-1 568.01 au和 E =-1 566.00 au,由此可见第一种构型能量最低,是最稳定的构型,并且经振动频率分析所得分子结构无虚频,即为稳定分子构型,因此我们将其具体的结构参数列于表1中,进行分析探讨。
由表1的键长数据可以看出,奥利司他分子中C35—O40、O40—C41键长分别为0.146 9、0.134 1 nm,与未取代的碳氧单键键长0.143 0 nm相比,前者键长增加,后者键长减小;C41—C69、C69—C71键长为 0.153 1、0.154 6 nm,与一般碳碳单键键长0.154 0 nm相比,前者键长略小,后者略有增加;C69—N84、 N84—C86键长分别为0.145 4、0.136 5 nm,与未取代的碳氮单键键长0.147 0 nm相比,均减小。这些数据显示C35~C80这部分取代基在分子中形成了一个π共轭系统。
由图2可知,奥利司他分子并不是一个平面结构,从表1中的二面角数据可以看到,C1~C35所在的碳链处于x轴负方向,为平面结构。而与C35相连的两条支链则开始出现扭曲,∠C29—C32—C35—C37 =-69.72°,∠C32—C35—C37—C67=126.20°,∠C35—C37—C67—O46=-65.63°,∠C32—C35—O40—C41=-157.51°,说明C1~ C35所在碳链与C37~C67所在支链存在绝对值为60°左右的二面角,与O40—C41所在支链存在绝对值为157.51°的二面角;∠C67—C37—C35—O40=-115.36°,∠C37—C35—O40—C41=78.49°,说明C37—C67所在支链与O40—C41所在支链之间有绝对值为70°左右的二面角。
2.2 红外光谱及分析
中红外光谱图(400~4 000 cm-1)能反映分子转动和振动的特征,引起分子偶极矩变化的振动形式可分为伸缩振动和弯曲振动两种。伸缩振动包括对称伸缩振动和反对称伸缩振动,弯曲振动又包括剪式振动、对称变形振动、不对称变形振动、平面摇摆振动、垂直摇摆振动和扭曲振动[12-15]。本文在优化稳定结构的基础上,利用密度泛函B3LYP/6-31G方法,计算得到了奥利司他分子在中红外区的红外振动光谱,如图4所示。分析振动频率发现,按照分子红外吸收峰情况,可将奥利司他红外振动光谱分为400~700、700~1 200、1 200~1 600、1 800~2 000、2 900~3 200 cm-1 5个区域进行分析。
在400~700 cm-1区域内,奥利司他分子的主要振动形式为分子中各基团碳氢键的面内弯曲振动。由图3可以看出,这一区域内出现的谱峰较其他区域要弱,最强峰出现在639 cm-1的位置,它主要是由N84—H85、C69—H70、C71—H72、C71—H73的平面摇摆振动引起的。除此之外,这一波数范围内大量分子的振动没有相应的谱峰出现,分析振动频率数据可知,这主要是由于这些振动模式的强度较弱,没有引起分子偶极矩发生明显变化而导致的。
700~1 200 cm-1区域内,有两个较强吸收峰出现在708、736 cm-1处,主要是由N84—H85的平面摇摆振动和H73—C71—H72的不对称变形振动引起的,振动模式见图4;这个区域内的次强峰出现在1 147、 1 157 cm-1的位置,主要是由C44所在的四元环及环上取代基中的碳氢面外彎曲振动引起的,见图5。
1 200~1 600 cm-1区域出现的两个最强峰,位置分别在1 253、1 303 cm-1处,其中1 253 cm-1处的吸收峰主要贡献是H38—C37—H39的不对称弯曲振动和C69—H70的平面摇摆振动,其振动模式如图6所示;1 303 cm-1处的吸收峰主要是由N84—C86的伸缩振动、N84—H85和C86—H87的平面摇摆振动以及H70—C69—H72的对称变形振动引起的。
1 800~2 000 cm-1区域的谱峰主要是分子内基团的伸缩振动引起的,与其他区域中谱线的强度相比,本区域出现的振动峰整体强度是整个分子中最强的,其中最强峰出现的位置是1 856 cm-1,这也是整个光谱中的最强峰,主要是由C86=O88的伸缩振动引起的,同时伴随着N84—H85和C86—H87的平面摇摆振动;这一区域的次强峰出现在1 963 cm-1处,这也是整个红外光谱中的次强峰,主要是由C44=O47的伸缩振动引起的。
在2 900~3 200 cm-1区域,红外谱峰主要对应奥利司他分子中各基团的伸缩振动。这部分的最强峰出现在3 039 cm-1处,主要是由C1—C29支链上 H—C—H的对称伸缩振动引起的,其振动模式见 图7;而这部分的次强峰是在3 093 cm-1处出现,这恰好是由C1—C29支链上H—C—H的不对称伸缩振动引起的。
2.3 核磁共振及分析
核磁共振氢谱(1H NMR)和碳谱(13C NMR)可以反映化合物的分子结构[16-18],其中1H NMR能反映H原子所处的化学环境,13C NMR则可以反映有机化合物的结构骨架信息。本文在分子结构全优化的基础上,采用规范不变原子轨道NMR=GIAO的方法,以四甲基硅烷TMS为内标,计算了奥利司他分子的核磁共振1H NMR和13C NMR化学位移数据,结果见表2。
在核磁共振氢谱中,化学位移数值的大小反映了所讨论氢原子核外电子云密度的大小,即氢原子核外s电子的电子云密度的大小。s电子的电子云密度越大,化学位移的数值越小,反之亦然[19-21]。由表2中数据可知奥利司他1H NMR谱中化学位移值最大的是87号氢原子,δH=7.442 8×10-6,这是由于与该氢原子相连的86号碳原子上面取代了氮原子和氧原子,均为电负性强的基团,这些基团吸引电子,使键电子更靠近C86,因而对于相连的H87有去屏蔽作用,使其化学位移数值增大。其次是H68和H36,δH分别为4.604 9×10-6、4.019 4×10-6,二者化学环境相似,在其邻位碳上都有O的取代,吸引电子,对H68和H36有去屏蔽作用,使其化学位移值较大,并且数值相近。
由于有机化合物骨架是由碳原子构成的,有些官能团不含氢原子,但含碳原子,并且核磁共振碳谱的化学位移数值变化范围远远大于氢谱的,其谱线是一条一条呈现的,很少有谱线重叠的情况,因此13C NMR谱对于推导未知物结构或者确认结构都非常重要[19-21]。由表2的理论计算结果可知,奥利司他分子碳谱中化学位移值最大的3个碳:C41、C44和C86,其δC分别为161.21×10-6、155.24×10-6、137.88×10-6,这3个碳均为羰基碳,羰基的谱峰处于在13C NMR谱中的最低场,很容易识别。而羰基若与杂原子相连会产生比较大的高场位移,这就是C86的化学位移比其他两个羰基小的原因。化学位移在65.34×10-6和60.97×10-6的两个位置上的是C35和C67,二者均为与氧相连的次甲基,由于C67是处于四元环中的碳,因此其化学位移较C35略小。
3 结 论
本文采用密度泛函理论B3LYP方法,从理论角度研究奥利司他药物分子。选取6-31G(d,p)基组,对奥利司他分子的结构进行了最低能量优化,分析了其优化构型的特点,发现奥利司他分子不是一个平面结构,分子中有一个π共轭系统。
采用同样的方法,计算得到了奥利司他分子红外振动光谱,分析发现:在400~700 cm-1区域内,主要振动形式为分子中各基团碳氢键的面内弯曲振动;700~1 200 cm-1区域内,主要是平面摇摆振动和不对称变形振动;1 200~1 600 cm-1区域主要是不对称弯曲振动和平面摇摆振动;1 800~2 000 cm-1区域的谱峰,主要是分子内基团的伸缩振动引起的,本区域出现的振动峰整体强度是整个分子中最强的; 2 900~3 200 cm-1区域,主要对应奥利司他分子中各基团的伸缩振动。
对奥利司他分子氢谱的研究发现,化学位移值最大的是87号氢原子;分子碳谱中化学位移值最大的3个碳是C41、C44和C86。
总的来说,本文的研究结果可为奥利司他分子结构的鉴定提供光谱解析方面的理论依据,简化化合物结构鉴定工作。
参考文献:
[1]钱磊,向华,尤启冬,等. 减肥药物最新研究进展[J].中国新药杂志,2007,l6(6):437-442.
[2]涂春联,李红胜. 黄连解毒汤治疗肥胖型2型糖尿病临床研究[J].中医学报,2015,30(5):644-646.
[3]NG M,FLEMING T,ROBINSON M,et al. Global,regional,and national prevalence of overweight and obesity in children and adults during 1980-2013:a systematic analysis for the global burden of disease study 2013[J].Lancet,2014,384:766.
[4]AHN S M,KIM H,JI E,et al. The effect of orlistat on weight reduction in obese and overweight Korean patients[J].Arch Pharm Res, 2014, 37(4):512-519.
[5]LI Y, CHEN C, MA Y, et al.Multi-system reproductive metabolic disorder:significance for the pathogenesis and therapy of polycystic ovary syndrome ( PCOS)[J].Life Sci,2019,228:167-175.
[6]黄秋菊,李玉兰. 奥利司他疗效研究及不良反应[J].现代医药卫生,2018,34(19):3025-3027.
[7]曹媛,李娜,贺苗. 奥利司他对肥胖2型糖尿病患者血糖和血脂的影响[J].西南国防医药,2019,29(5):586-588.
[8]闵敏,阮祥燕,赵越,等. 奥利司他综合干预对超重或肥胖型PCOS患者雄激素及糖脂代谢的影响[J].首都医科大学学报,2019,40(4):572-577.
[9]侯亚利,邓飞.膳食疗法联合奥利司他治疗内分泌失调性肥胖的效果[J].航空航天医学杂志,2019(1):72-73.
[10]鄢兵,刘廷华,胡海山,等. HPLC法同时测定减肥药中四种违禁减肥药物成分[J].现代科学仪器,2012(1):104-107.
[11] 魏强. 高效液相色谱分析三类保健食品中常见违禁药物的方法研究[D].南昌:南昌大学,2008.
[12]刘存海,张勇,柳叶,等. 盐酸克伦特羅的密度泛函理论研究[J].计算机与应用化学,2014,31(2):223-225.
[13]于建成,唐延林,常瑞等. 基于密度泛函的茶多酚分子EGCG和GCG的光谱计算[J].光谱学与光谱分析,2019,39(6):1846-1851.
[14]刘存海,张勇,刘芬芬,等.香兰素的密度泛函理论研究 [J].化学工程师,2012,26(12):18-20.
[15]蔡开聪,杜芬芬,刘佳等.Gaussian软件在红外光谱学教学中的应用[J].化学教育,2014,35(8):50-53.
[16]何伟平,黄菊,陈秒慧,等. 头孢氨苄的密度泛函研究[J].四川大学学报(自然科学版),2018,55(3):564-570.
[17]许旋,徐志广,罗一帆.紫杉醇的核磁共振谱及其分子几何构型的从头算研究[J].物理化学学报,2002,18(5):420-425.
[18]DITCHFIELD R. Self-consistent perturbation theory of diamagnetism: I. a gauge-invariant LCAO method for NMR chemical shifts[J]. Mol. Phys.,1974,27:789.
[19]宁永成. 有机波谱学谱图解析[M].北京:科学出版社,2010.
[20]张华. 有机结构波谱鉴定[M].大连:大连理工大学出版社,2009.
[21]梁小蕊,牛妍懿,刘洁.海洋天然产物Trichodermaxanthone分子光谱的理论研究[J].海军航空工程学院学报,2019,34(5):459-464.
