Monoacylglycerol lipase reprograms lipid precursors signaling in liver disease
2020-08-18MatteoTardelli
Matteo Tardelli
Abstract Dietary oversupply of triglycerides represent the hallmark of obesity and connected complications in the liver such as non-alcoholic fatty liver disease and non-alcoholic steatohepatitis, which eventually progress to cirrhosis and hepatocellular carcinoma. Monoacylglycerol lipase is the last enzymatic step in the hydrolysis of triglycerides, generating glycerol and fatty acids (FAs), which are signaling precursors in physiology and disease. Notably, monoacylglycerol lipase (MGL) also hydrolyzes 2-arachidonoylglycerol, which is a potent ligand within the endocannabinoid system, into arachidonic acid - a precursor for prostaglandin synthesis; thus representing a pivotal substrates provider in multiple organs for several intersecting biological pathways ranging from FA metabolism to inflammation, pain and appetite. MGL inhibition has been shown protective in limiting several liver diseases as FAs may drive hepatocyte injury, fibrogenesis and de- activate immune cells, however the complexity of MGL network system still needs further and deeper understanding. The present review will focus on MGL function and FA partitioning in the horizons of liver disease.
Key words: Lipid metabolism; Monoacylglycerol lipase; Non-alcoholic steatohepatitis; Non-alcoholic fatty liver disease; Cannabinoid; Nuclear receptors
INTRODUCTION
Dietary lipids are major sources of energy in the body and serve a variety of vital functions[1]. They represent pivotal substrates for beta oxidation, fueling the production of cellular energy as well as being precursors of all lipid classes, including prostaglandins, steroid hormones and those that form biological membranes[1]. Notwithstanding their key roles, aberrant lipid metabolism may become harmful to cells, as free fatty acids (FAs) are known to be powerful signaling molecules[2,3]. FAs and their metabolites regulate several intracellular processes such as gene transcription and expression, post-transcriptional/translational modification of proteins, also directly modulating an array of enzyme activities as co-activators[4,5]. Fat stores in the adipose tissue (AT) are the major energy reserves in mammals. Upon food intake, lipid species are absorbed in the intestine and delivered to the circulation; the surplus of dietary non-esterified fatty acids (NEFAs) which are not immediately needed by the body, are re-esterified into triglycerides (TG) that are subsequently stored in cytosolic lipid droplets of the adipocytes[3]. During fasting for instance, these TG stores in the AT can be released by hydrolytic cleavage, and the resulting NEFAs reach peripheral tissues such as liver and muscle to be processed for β-oxidation and therefore energy/ATP production[6]. Of interest, also non-adipose organs can effectively esterify NEFAs into TG and re-hydrolyze them back into NEFAs according to the energy demands[6]. Excessive amounts of TGs store into AT and other organs such as liver leading to lipotoxicity as well as non-alcoholic fatty liver disease and type 2 diabetes[7]. The cleavage of primary/secondary ester bonds between long chain FAs and glycerol backbone that form TGs, depends on specific hydrolases commonly called neutral lipases (to be differentiated from lysosomal acid lipases)[3,8]. Three main enzymes are involved in the thorough hydrolysis of TGs in cellular lipid stores: adipose triglyceride lipase exclusively performs the first step hydrolyzing TGs to generate diacylglycerols (DGs) and FAs[3]. Hormone-sensitive lipase instead is capable of hydrolyzing several acylesters including TGs, DGs, and monoacylglycerols (MGs)[3,6]. Finally, monoacylglycerol lipase (MGL) efficiently cleaves MGs into glycerol and FAs[9](Figure 1). This process is aided by comparative gene identification-58 (CGI-58, also known as ABHD5), which interacts with perilipin, protecting or exposing the TG core of a lipid droplet to lipases (mainly adipose triglyceride lipase)[10]and therefore enhancing their activity[11]. Hydrolysis is finely balanced by concomitant reesterification of MGs/DGs into TGs, which is performed by monoacyglycerol acyltransferases (MGAT) and diacylglycerol acyltransferase (DGAT) respectively, as shown in Figure 1.
MGL is a particularly interesting enzyme as substantially bridges organ-specific nutrient metabolism to central and peripheral endocannabinoid and eicosanoid systems[12]. The fact that the latter holds such a multipurpose and promiscuous role in several physiological processes is driving a resurgence of interest in this lipase as putative drug target for several different disorders[13]. Although, very well characterized in behavioral studies and central nervous system disease models[14-16](in connection to the endocannabinoids and prostaglandins network), the contribution of MGL to metabolic liver disease, cholestasis, inflammation, and fibrosis is surprisingly unknown.
PRE-CLINICAL STUDIES ON MONOACYLGLYCEROL LIPASE DELETION/ INHIBITION

Figure 1 Catabolic pathways regulating triglycerides levels in adipocytes. Adipose triglyceride lipase, hormone sensitive lipase, and monoacylglycerol lipase act on triglycerides in a series of subsequent reactions in order to generate glycerol and fatty acids. Comparative gene identification-58 binds to perilipin facilitating lipolysis, as a known activator of adipose triglyceride lipase. Intermediates of this catabolic pathway may also be re-esterified to phospholipids and triglycerides by the enzymes monoacyglycerol acyltransferases and diacylglycerol acyltransferase. ATGL: Adipose triglyceride lipase; HSL: Hormone sensitive lipase; MGL: Monoacylglycerol lipase; TGs: Triglycerides; FAs: Fatty acids; CGI-58: Comparative gene identification-58; MGAT: Monoacyglycerol acyltransferases; DGAT: Diacylglycerol acyltransferase.
MGL is the rate-limiting enzyme of MG degradation that derive from intra- /extra- cellular phospholipids or TGs[17]. The extracellular most important source of MGs is TG-rich lipoproteins such as the liver-derived very low density lipoproteins[18]. TGs are released from circulating very low density lipoproteins by the action of the endothelial lipoprotein lipase that generates FAs, hydrolyzing TGs in positions sn-1- and sn-3, thus generating MG species[19]. Digestion of dietary TG is also aided by pancreatic lipase that can generate in addition fair amounts of MGs[20]. Extracellular derived MGs are internalized by cells and may be further degraded by MGL or alternatively reesterified to TGs by the enzymatic activities of MGAT and DGAT (Figure 1). In fact, MGAT and DGAT are key players in enterocytes and particularly in the small intestine, where they are thought of synthesizing up to 80% of the TG incorporated into chylomicrons[21]. In addition, another source of MGs can directly originate from the intracellular TGs storage in cytosolic lipid droplets[3].
MGL is a pivotal lipase that produces signaling lipid, notably hydrolyzing 2-AG (which is a known ligand of cannabinoid receptors) into arachidonic acid (AA)[9]. Given the multiple roles of the cannabinoid system in regulation of several key processes in human physiology including (but not limited to) appetite, pain and even cancer, enzymes targeting the cannabinoid catabolism are potentially interesting candidates for drug discovery and pharmaceutical development[14]. MGL is ubiquitous in the body and its expression varies greatly in an organ-specific fashion, being present in brain and in peripheral tissues such as the kidney, testis, ovary, adrenal gland, AT, and heart. At the cellular level, MGL mainly localizes in the cytoplasm as well as in the plasma membrane and in the lipid droplet[22]. Interestingly, molecular strategies exploiting genetic or pharmacological inactivation of MGL in mice resulted in a significant accumulation of MGs in several tissues demonstrating its central role in MG catabolism[9]. In diet induced obesity models, MGL ablation showed to be protective in the development of glucose intolerance and insulin resistance, although reduced MGL activity was partially reverted by hormone-sensitive lipase[23]. This phenotype was shown to be influenced by the effects of MGL on the central nervous system and derived behavioral changes triggered by the endocannabinoid pathway[23]. Importantly, MGL knockout mice (Mgl-/-) showed neuroprotective properties in a parkinsonian mouse model, with consequent reduction of multiple prostaglandins and eicosanoids mediators in the brain, including prostaglandin -E2, -D2, -F2, and thromboxane B2[24]. In keeping with this parkinsonian mouse models, MGL´s network of endocannabinoid and prostaglandin regulation was also demonstrated to contribute to the pathogenesis of Alzheimer´s disease, representing a promising target for its prevention/treatment[16]. Chronic inactivation of MGL in the central nervous system was shown to lead to a 2-AG oversupply and consecutive desensitization of cannabinoid 1 receptor (CB1r) signaling[23]. Intriguingly, central CB1r inactivation was correlated to a higher incidence of depression and several other psychiatric side effects in patients[25], which led to consequent withdrawal of the anti-obesity drug rimonabant, an inverse CB1r agonist[23]. Experiments with CB1r/MGL double-KO mice revealed that oral but not intraperitoneal lipid administration strongly suppressed the appetites ofMgl-/-and CB1r/MGL DKO mice, but not affecting wild-type and CB1r-/-mice[26]. In this study, appetite suppression was reversed by vagotomy, suggesting a mechanistical involvement of MGL in the gut-brain axis regulation of appetite[26]. In keeping, administration of an MGL inhibitor called MJN110, reduced food intake in rats[27]. Notably, pharmacological blockade of MGL with JZL184 caused increased systemic 2-AG levels in a mouse model of myocardial infarction[28]. This resulted in elevated cardiac C-X-C motif chemokine ligand-1, -2, and matrix metallopeptidase-9 levels as well as increased cardiac neutrophil/monocyte counts 24 h after infarction compared to vehicle-treated mice[28]. In another study, dietary supplementation of linoleic acid in a range of 1% to 8% significantly increased 2-AG in the liver and small bowel, suggesting possible dietary tools to modulate endocannabinoid ligands[29].
Of interest, a recent study characterized a unique population of human bone marrow adipocytes[30], discovering a profound downregulation of MGL expression which was shown to underlie the metabolic differences in the phenotype/lipid metabolism, also justifying bone marrow adipocytes’ resistance to caloric restrictions[30]. Further studies showed that whenMgl-/-animals were challenged with high fat diet feeding, great amounts of 2-AG were detected in the brain, although affecting neither food consumption nor body weight, therefore evidencing cannabinoid receptors desensitizing effects[23]. Notably, the liver content of certain species of saturated and unsaturated MGs were highly enriched inMgl-/-, showing after 12 wk high fat diet better insulin sensitivity and glucose tolerance[31,32]; although whether adipose depots or skeletal muscle were the main players in determining this phenotype, was not thoroughly investigated. Several other works evidenced a role of MGL in tumor growth/metabolism as well as oncogenic signaling[33], being associated with gastrointestinal stromal tumors[34]and hepatocellular carcinoma (HCC); others instead showed that MGL deletion resulted into significant colorectal cancer growth inhibition[35]while also reducing ischemia/reperfusion-induced lung injury and inflammation[36,37]. Additionally, MGL was found to be part of a gene signature determining stem-like properties of cancer cells in prostate cancer[38]; this further supported a role for this enzyme in pro-tumorigenic metabolism, involving the dual control of endocannabinoid and FA pathways[38,39].
Xianget al[40]elegantly showed that MGL deficiency caused lipid overload in tumor associated macrophages[40]. Intriguingly, MGL expression in macrophages was strongly decreased in cancer tissues and positively correlated with the overall survival of cancer patients[40]. Mechanistically, MGL deficiency was shown to promote CB2r/TLR4 axis and subsequent macrophage activation, which in turn suppressed the function of tumor associated CD8+ T cells. In addition, treatment with CB2r antagonist drugs delayed tumor progression in inoculated and genetic cancer models[40].
In the liver, MGL ablation was demonstrated to attenuate LPS-induced inflammation whilst whole body genetic and pharmacological inhibition protected against inflammation and liver lesions provoked by ischemia/reperfusion injury[41]. Intriguingly, hypoxia training was demonstrated to induce MGL expression and ameliorate hepatic steatosis in obese mice, showing decreased 2-AG levels and expression of CB1r[42].
2-AG accumulation in the liver has been shown to have anti-inflammatory and antifibrotic effects through CB2r signaling, conversely CB1r activation associates with increased liver damage and fibrosis in different models of liver injury[43](Figure 2). Habibet al[43]suggested that the increased 2-AG levels following MGL blockade selectively stimulate CB2r but not CB1r, thus resulting in hepato-protective effects[43]. In their study, lack of MGL prompted fibrosis regression due to autophagy-mediated anti-inflammatory mechanisms in macrophages[43]. In details, using a mouse model lacking MGL in the myeloid lineage (MglMye-/-), authors demonstrated that MGL inhibition in immune cells is sufficient to reduce hepatic fibrosis and inflammation after carbon tetrachloride (CCL4) injection[43]. In line with what Caoet al[41]previously showed, that systemic MGL inhibition had protective effects on hepatocytes in different models of acute liver injury. These evidence was dependent on the enhanced CB2r activation and modulation of eicosanoid pathways, resulting into diminished neutrophil infiltration and neutrophil-mediated liver damage[41].
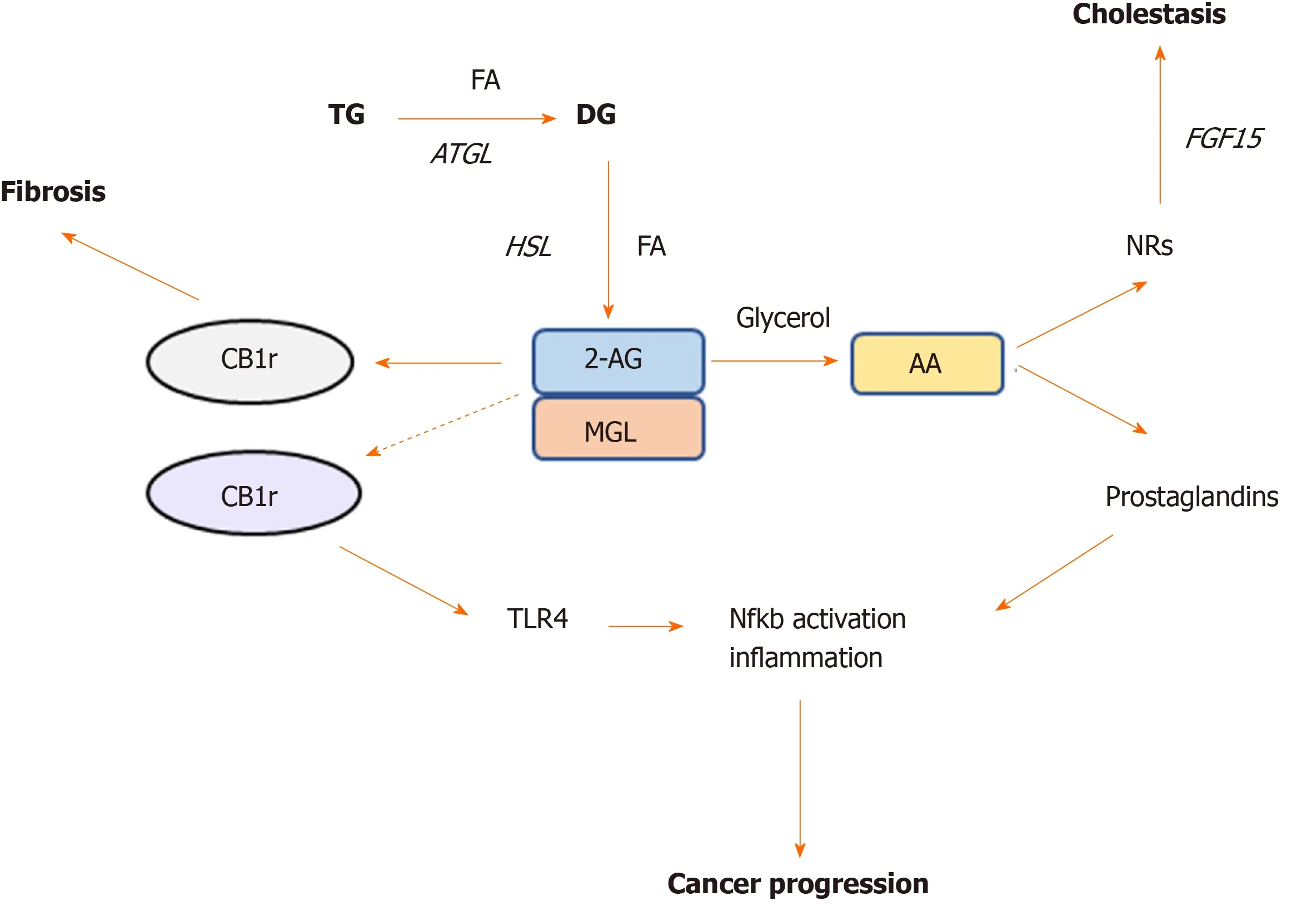
Figure 2 Monoacylglycerol lipase represent a crossroad between cannabinoid and lipid signaling pathways. Monoacylglycerol lipase is the key enzyme degrading the endogenous cannabinoid ligand 2-AG, which in turn is able to bind either cannabinoid receptor (CB) -1r or -2r. Activation of CB1r was found to promote fibrosis whereas CB2r is involved in TLR4 activation and immune cells recruitment in cancer. Monoacylglycerol lipase action further hydrolyzes 2-AG into arachidonic acid, which is a precursor of prostaglandin synthesis, the main drivers of inflammation. Arachidonic acid was also found to bind nuclear receptors such as farnesoid X receptor and peroxisome proliferator activated receptors in the intestine and ameliorate cholestatic disease. AA: Arachidonic acid; NRs: Nuclear receptors; CB: Cannabinoid; MGL: Monoacylglycerol lipase; HSL: Hormone sensitive lipase; ATGL: Adipose triglyceride lipase; TG: Triglyceride; FA: Fatty acid.
In a microarray study, the expression of MGL was shown to be enriched in HCC tumors than in matched non-tumor tissues[44]. The upregulation of MGL in HCC cells promoted cell growth and invasion through pro-tumorigenic lipid signaling[44]. Importantly, MGL was demonstrated to facilitate HCC progressionvianuclear factor kappa light chain enhancer of activated B cells-mediated epithelial-mesenchymal transition[44]. A recent work from our group strongly demonstrated a central role of MGL inhibition in the pathophysiology of cholestatic liver disease[45]. Using total body knockout mice challenged with cholestatic diet [3,5-diethoxycarbonyl-1,4-dihydrocollidine (DDC)] and pharmacological inhibition (JZL184) of MGL in Mdr2 (Abcb4) knock out -Mdr2-/-(an established mouse model that develops spontaneous cholestasis[46]), we evidenced beneficial effects in sclerosing cholangitis development and resolution[45]. We showed that both mouse models were protected from cholestatic liver disease due to a crosstalk mechanism involving the gut-liver axis, microbiome modulation and substrate (AA) accumulation in the intestine (Figure 3). Notwithstanding that AA is a known precursor of pro-inflammatory mediators, in this study it was demonstrated to bind nuclear receptors such as peroxisome proliferator activated receptor alpha and gamma (PPAR-α, -γ) and farnesoid X receptor diminishing intestinal inflammation and consequently impacting liver bile acid synthesisviafibroblast growth factor 15[45]. Nevertheless, the development of tissue specific and inducible knockout models would deliver better mechanistic understanding on which organ is playing the most relevant role in the development of cholestasis and other liver diseases after MGL invalidation.
Although several pre-clinical studies showed promising/beneficial effects of MGL blockade in multiple disease models, inhibition of this key metabolic enzyme requires cautious evaluation. On the one hand, the MGL inhibitor named JZL184 (firstly characterized by Longet al[47]) has been shown to have low specificity and thus blocking activities of other hydrolases such as fatty acid amide hydrolase and carboxylesterases. On the other hand, another inhibitor called MJN110 was shown to have less activity towards other serine hydrolase systems, also being more effective at enhancing 2-AG levels in the brain than JZL184[48,49]. In keeping with this evidence, both inhibitors are currently used in preclinical studies andin vitrosystems to explore the effects of MGL inhibition. In a recent work, a new candidate JNJ-42226314 was identified as a reversible and highly selective MGL inhibitor and several other approaches reported developments of positron emission tomography radioligand inhibitors for MGL such as PF-06809247[50,51].
In summary, MGL appears to serve as key metabolic hub for several pathways within chronic liver disease such as sclerosing cholangitis, non-alcoholic fatty liver disease, fibrosis and HCC which are disorders affecting complex network of several cell types and molecular mechanisms.
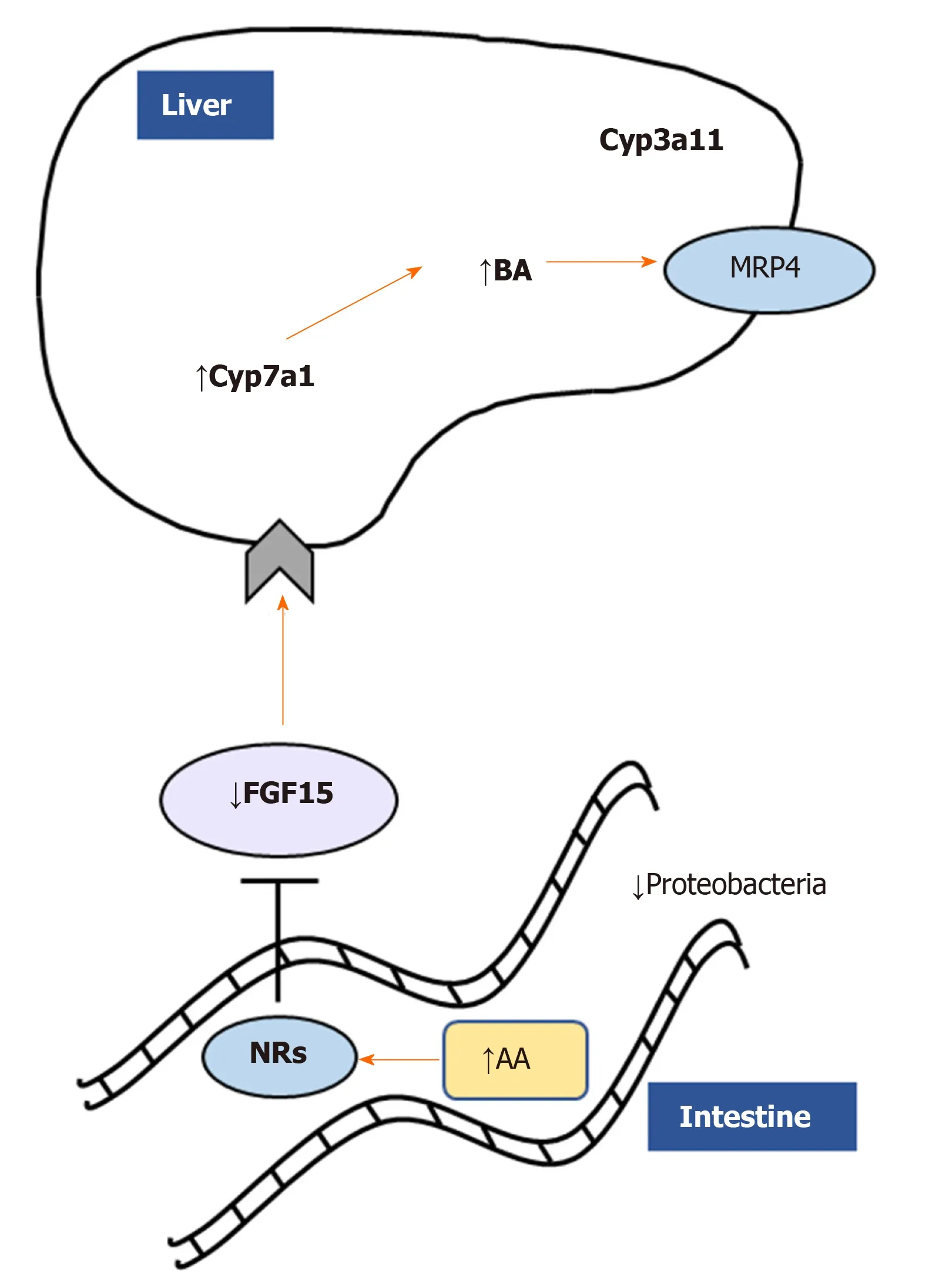
Figure 3 Monoacylglycerol lipase deletion impacts gut-liver axis via nuclear receptor and microbiome modulation. Monoacylglycerol lipase ablation ameliorates cholestatic liver disease induced by 3,5-diethoxycarbonyl-1,4-dihydrocollidine challenge diminishing fibrosis, inflammation, and fatty acid metabolism/oxidation in the liver. Accumulation of arachidonic acid binds nuclear receptors such as farnesoid X receptor, downregulating in turn fibroblast growth factor 15 and inducing bile acids synthesis and detoxification as shown by Cyp7a1/Cyp3a11. In addition, proinflammatory Proteobacteria were diminished in feces from Mgl-/- mice. AA: Arachidonic acid; NRs: Nuclear receptors; FGF15: Fibroblast growth factor 15; BA: Bile acids.
CLINICAL RELEVANCE OF MONOACYLGLYCEROL LIPASE AND ITS GENETIC MUTATION
Only a handful of studies explored the clinical relevance of MGL in human patients, and those mainly focused on rare genetic variants known as single nucleotide polymorphism (SNPs), which may in turn influence determinant lipid-related traits and pathologies[52]. Intriguingly, 4 MGL SNPs named rs13076593, rs782440, rs541855, rs549662 were associated with increased LDL particle size, whilst the rs3773159 with type 2 diabetes[53]. Moreover, 20 common variants of theMGLgene were associated with high BMI in a cohort of 289 individuals (of which 147 controls and 142 extremely obese)[54]. Interestingly, the genotype rs604300 was shown to have protective effects against childhood abuse-related increases in cannabis dependence and was suggested to relate to epigenetic modulation of MGL expression[55]. Lastly, the variant Pro129Thr of one of the endocannabinoid inactivating enzyme called fatty acid amide hydrolase was found to significantly associate with both street drug use and problematic drug/alcohol use[56]. However, this could not be replicated in another study in which no associations were found between Pro129Thr or other MGL SNPs and alcoholism in 729 Japanese patients with alcoholism and 799 healthy controls[57].
CONCLUSION
Further work is needed to understand in detail the specific tissue/cell contribution to the beneficial effects of MGL deletion observed in many studies and disease types. Of special interest would be unveiling the role of MGL in complex cancers such as HCC and cholangiocellular carcinoma or other liver conditions such as alcoholic liver disease, as no data are available yet. Moreover, novel therapeutic avenues may explore functional antagonism of MGL oni.e., the endocannabinoid system, which could be implemented with temporary approaches such as reversible blockade, small molecules, or immuno-therapy systems to study short-term efficacy. This could ultimately lead to better speculations and understanding of the complex network of the lipid machinery involved in the development of liver disease, insulin resistance and type 2 diabetes paving the way to novel pharmacological treatments.
杂志排行

World Journal of Gastroenterology的其它文章
- Chinese expert consensus and practice guideline of totally implantable access port for digestive tract carcinomas
- Type I and type II Helicobacter pylori infection status and their impact on gastrin and pepsinogen level in a gastric cancer prevalent area
- Retrievable puncture anchor traction method for endoscopic ultrasound-guided gastroenterostomy: A porcine study
- Predictors of irreversible intestinal resection in patients with acute mesenteric venous thrombosis
- Multiphase convolutional dense network for the classification of focal liver lesions on dynamic contrastenhanced computed tomography
- Chronic atrophic gastritis detection with a convolutional neural network considering stomach regions