含多官能团Gemini表面活性剂的合成及性能
2020-08-17王丽艳杨佳李云晴唐丽刘家庆杜昱嬴吴定成
王丽艳,杨佳,李云晴,唐丽,刘家庆,杜昱嬴,吴定成
(齐齐哈尔大学化学与化学工程学院,黑龙江齐齐哈尔161006)
Gemini 表面活性剂是一类具有两亲水基、两条疏水长链和一个连接基团的双亲基的表面活性剂[1-2]。与传统表面活性剂相比,它具有更优异的表面活性,可用作洗涤剂[3]、杀菌剂[4]、浮选捕收剂[5]及乳化剂[6]等。在油田[7-8]、金属缓蚀[9]以及无机材料合成[10]等领域被广泛应用。
目前国内外已报道合成的酰胺型双子表面活性剂主要集中在季铵盐型双子表面活性剂的研究。Akbas 等[11]合成了一系列含有酰胺键的季铵盐阳离子双子表面活性剂,研究结果表明该类双子表面活性剂的CMC 明显低于单子表面活性剂;Zhang 等[12]合成一系列含有酰胺键的季铵盐阳离子双子表面活性剂,研究发现该类表面活性剂具有较高的表面活性;Hu 等[13]合成一系列含有酰胺键刚性连接基的双子季铵盐表面活性剂,研究了胶束形成的热力学参数,结果表明胶束形成的过程是自发、放热的熵驱动过程;Yang等[14]合成了一系列含酯键的季铵盐双子表面活性剂,结果表明该类型表面活性剂比传统的表面活性剂具有更低的CMC,较好的黏附性等性能。仅含酰胺键与同时含酯键和酰胺键的构象相比柔性差,且在空气/水界面占有的面积更大[15],不利于胶束形成,表面活性较差;而研究报道鲜见亲油基中同时含酰胺键和酯键的Gemini 阳离子表面活性剂。因此,在季铵盐Gemini 表面活性剂中同时引进酰胺键和酯键,有利于提高表面活性剂的表面活性。
本文以来源易得的原料合成既含酯键又含酰胺键的季铵盐Gemini 表面活性剂系列,对其表面性能进行研究,并探讨该类表面活性剂的泡沫性和乳化性,与仅含有酰胺键的Gemini表面活性剂[16]进行了对照,为实际应用提供理论研究依据。
1 实验材料和方法
1.1 实验试剂与材料
长链伯胺(碳链长度为8、12、14 和16)、间苯二甲酰氯、碳酸钾,北京伊诺凯科技有限公司;氯乙酰氯,上海谱振生物科技有限公司;N,N-二甲基乙醇胺,天津市科密欧试剂有限公司;丙酮、乙腈、乙酸乙酯、无水乙醇、苯、二甲苯,天津市凯通化学有限公司;液体石蜡,北京化工厂;以上均为分析纯;机油,胜牌(上海)化学有限公司。
1.2 实验仪器
AVance 600 型超导核磁共振波谱仪,瑞士Bruker 公司;MPA100 型全自动熔点仪,美国SRS公司;Nicolet 750型傅里叶变换红外光谱仪,美国PE 公司;电子恒温水浴锅,天津市泰斯特仪器有限公司;DDS-307 型电导率仪,上海雷磁仪器厂;Krüss K100型表面张力仪,德国Krüss公司。
1.3 实验方法
1.3.1 中间体氯乙酰烷基胺(A)的合成
烷基伯胺和氯乙酰氯反应得到中间体A[17],合成路线如图1所示。

图1 中间体A的合成路线
以合成氯乙酰十二烷基胺A2为例,在100mL三口瓶中依次加入正十二烷基胺(1.854g,10mmol)、碳酸钾(0.8293g,6mmol)、三氯甲烷(20mL),冰水浴条件下,用恒压滴液漏斗缓慢滴加 氯 乙 酰 氯(1.355g, 12mmol) 与 三 氯 甲 烷(15mL)的混合溶液,约30min 滴加完毕。用薄层色谱法(TLC)监测反应进程,反应2.5h,停止反应,使用旋转蒸发仪除去溶剂。用无水乙醇对粗产物进行重结晶,得到2.042g白色固体中间体A2,产率78.00%,熔程63~66℃。用上述方法得到其他3个中间体A1、A3和A4,具体物理参数如下,A1为白色固体,产率80.10%,熔程52~54℃;A3为白色固体,产率81.30%,熔程71~74℃;A4为白色固体,产率80.00%,熔程75~79℃。
1.3.2 中间体二(N,N-二甲基胺基乙基)间苯二甲酸酯(B)的合成
间苯二甲酰氯和N,N-二甲基乙醇胺反应得到中间体B[18],合成路线如图2所示。

图2 中间体B的合成路线
向100mL 三口瓶中依次加入N,N-二甲基乙醇胺(1.783g,20mmol)和丙酮(20mL),在氮气保护冰水浴条件下,用恒压滴液漏斗将间苯二甲酰氯(1.015g,5mmol)和丙酮(20mL)混合液匀速滴入反应装置中,30min 滴加完毕,TLC 跟踪反应进程,10h 后停止反应,除去溶剂丙酮,得到1.211g无色透明的油状液体,产率为80.71%。
1.3.3 目标产物的合成
中间体A与中间体B反应得到目标产物I系列,合成路线如图3所示。
以化合物I12合成为例,向三口瓶中依次加入中间体氯乙酰十二烷基胺(A2,0.5236g,2.2mmol)、中间体B(0.3000g,1mmol)、乙腈(30mL),在75~80℃下反应,TLC 监测反应进程,反应5.5h,停止反应。旋蒸除去溶剂,冷却得到固体粗产物,用乙酸乙酯对粗产物进行重结晶,得到0.8370g 白色固体粉末,产率为88.60%,熔程为166~168℃。
用上述方法合成目标产物I8、I14、I16,具体物理参数如下,I8为白色固体,产率88.70%,熔程165~167℃;I14为白色固体,产率88.50%,熔程168~170℃;I16为白色固体,产率88.10%,熔程169~171℃。
1.4 性能测试
(1) 电 导 率 用DDS-307 电 导 率 仪,在298.15K、308.15K、318.15K、328.15K 下,采用稀释法[19],分别测试目标产物从高到低不同浓度水溶液的电导率值,绘制κ-C曲线。
(2)表面张力 在298.15K下,用吊环法分别测量不同浓度目标产物水溶液的表面张力值,绘制γ-c曲线。
(3)泡沫性能 按照文献[20],分别配制0.1%的待测的目标产物溶液,在25℃的恒温水浴锅中进行测量。取20mL 溶液置于100mL 具塞量筒中,在恒温水浴锅中恒温5min,上下剧烈振荡25 次,放回水浴锅中并立即按下秒表,记录初始泡沫高度(H1,cm)和5min后泡沫高度(H2,cm),稳泡性的计算见式(1)。

(4)乳化性能 按照文献[21]分别配制质量分数为0.1%的目标产物水溶液,以液体石蜡为例,量取40mL 目标产物水溶液置于100mL 具塞量筒中,再加入40mL 液体石蜡混合,上下猛烈振荡5次,静止60s,重复5 次,用秒表计时,记录分出10mL水所用时间。
2 结果与讨论
2.1 结构表征
2.1.1 FTIR分析
对目标产物(I8、I12、I14、I16)采用KBr压片法进行红外光谱(FTIR)测定,产物的FTIR 谱图如图4所示。
由图4可见,以I12为例进行分析,FTIR(KBr,v/cm-1):3424cm-1处为酰胺键中的N—H 键伸缩振动吸收峰,3030cm-1为苯环上C—H 伸缩振动吸收峰,2929cm-1和2852cm-1处分别为—CH3和—CH2—的对称和反对称伸缩振动吸收峰,1724cm-1处为酯键中羰基的伸缩振动吸收峰,1672cm-1处为酰胺键中羰基的伸缩振动吸收峰,1550cm-1处为酰胺键中N—H面内弯曲振动吸收峰,1463cm-1处为—CH3弯曲变形振动吸收峰,1222cm-1处为C—N 伸缩振动吸 收 峰, 1087cm-1处 为C—O—C 伸 缩 振 动,733cm-1处为长烷基链—(CH2)n—的弯曲振动吸收峰。FTIR谱图证明了目标产物中含有苯环、N—H键、C—H 键、羰基、—CH3、—CH2—、C—N和—(CH2)n—。
2.1.21H NMR分析
以I12的1H NMR谱图为例,如图5所示。
中间体A2:1H NMR(600MHz,CDCl3)δH:6.56(br s,2H,—NH—), 4.05 (s, 2H,—CH2—Cl), 3.29~3.32 (m,2H,—NH—CH2—),1.52~1.57[m,2H,—CH2—(CH2)9—),1.26~1.31[m,18H,—(CH2)9—], 0.87~0.89 (t, 3H,J=7.2Hz,—CH3)。
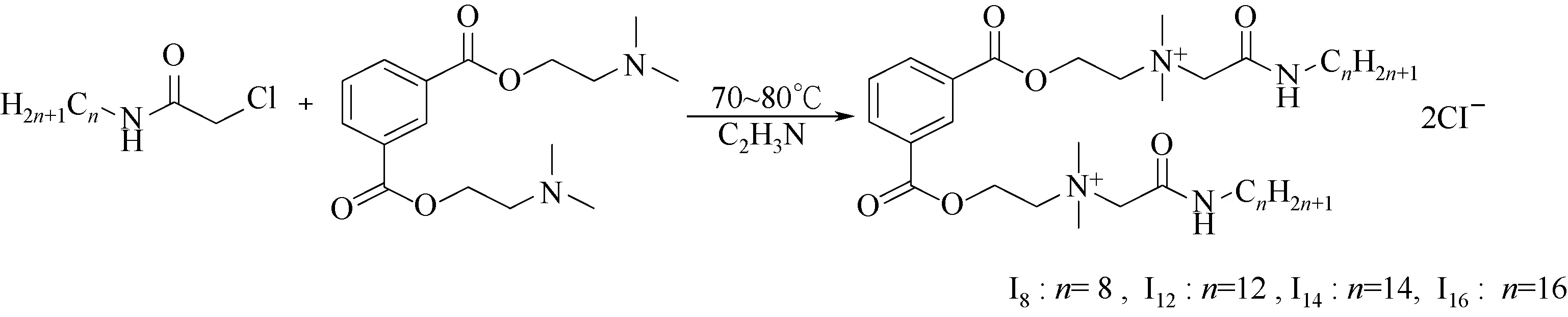
图3 目标产物I的合成路线

图4 I8、I12、I14和I16的FTIR谱图
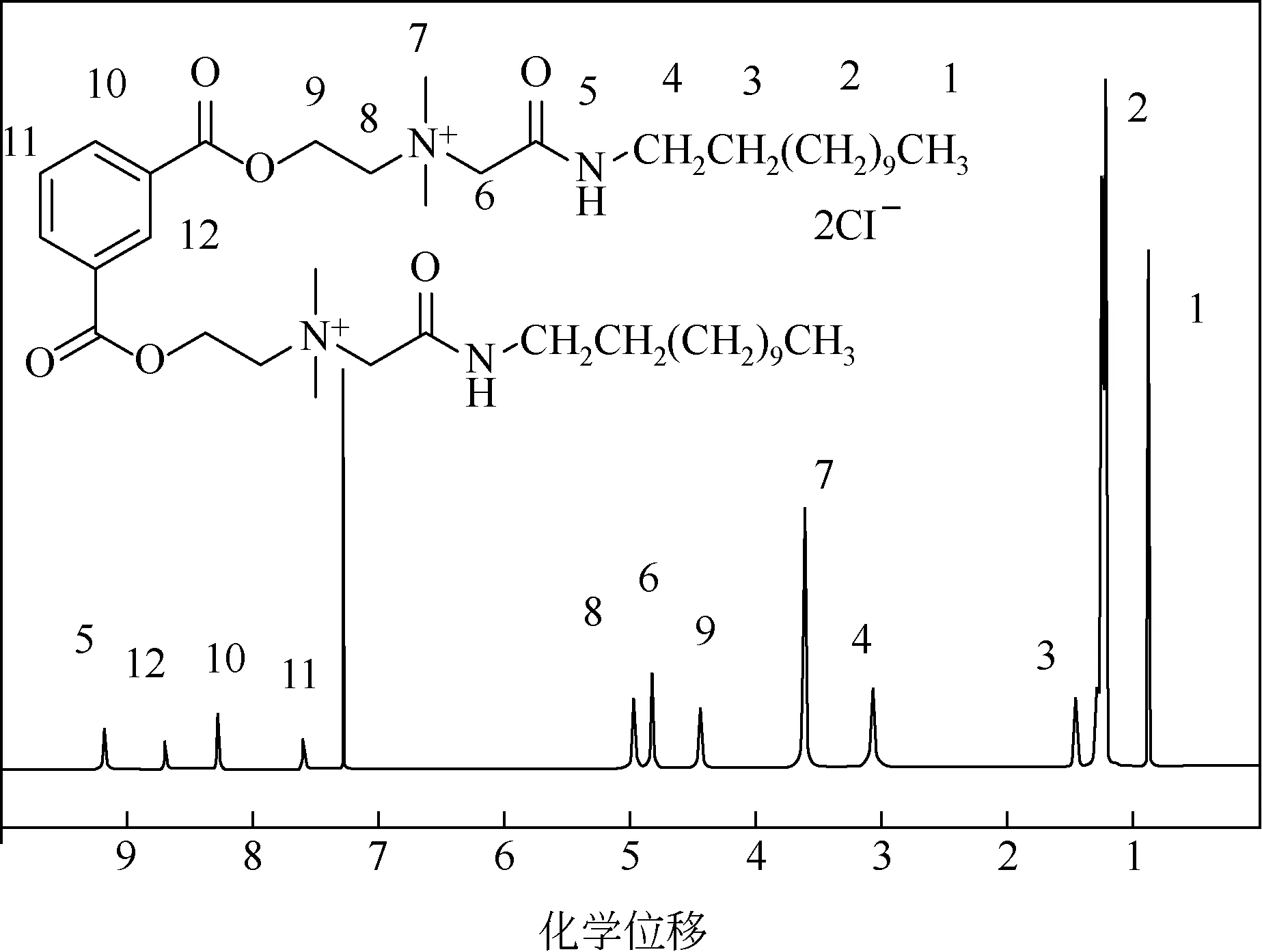
图5 I12的1H NMR谱图
中间体B:1H NMR(600MHz,CDCl3)δH:8.70(s,1H, PhH), 8.23 (d, 2H,J=7.8Hz, PhH), 7.52~7.54 (t,1H,J=7.8Hz, PhH), 4.45~4.47 (t,4H,J=6.0Hz,—O—CH2—), 2.73~2.75 (t, 4H,J=5.7Hz, —CH2—N—),2.35[s,12H,N—(CH3)2]。
目标产物I8:1H NMR(600MHz,CDCl3),δH:9.22(br s,2H,O==C—NH—),8.41(s,1H,PhH),8.27~8.28(m, 2H, PhH), 7.58~7.61 (t, 1H,J=7.8Hz, PhH), 4.95~4.96 (t, 4H,J=2.7Hz, —O—CH2—CH2—N+—),4.83 (s, 4H, —N+—CH2—C==O), 4.42~4.43 (t, 4H,J=2.7Hz, —O—CH2—CH2—N+—), 3.60 [s, 12H,—N+—(CH3)2], 3.06~3.09 (m, 4H, —NH—CH2—),1.45~1.48 (m, 4H, —NH—CH2— CH2—), 1.23~1.28 [m, 20H, — (CH2)5—], 0.85~0.88 [t, 6H,J=6.9Hz,—(CH2)5—CH3]。
目 标 产 物I12:1H NMR (600MHz, CDCl3),δH:9.19~9.20 (t, 2H,J=5.1Hz, O==C—NH—), 8.68 (s, 1H,PhH), 8.26~8.27 (m, 2H, PhH), 7.57~7.60 (t, 1H,J=7.8, PhH), 4.95 (br s, 4H, —O—CH2—CH2—N+—),4.80 (s, 4H, —N+—CH2—C==O), 4.44 (br s, 4H,—O—CH2—CH2—N+—),3.61[s,12H,—N+—(CH3)2],3.06~3.10 (m, 4H, —NH—CH2—), 1.45~1.48 (m, 4H,—NH—CH2—CH2—),1.23~1.30 [m, 36H,—(CH2)9—],0.87~0.90[t,J=7.2Hz,6H,—(CH2)9—CH3]。
目 标 产 物I14:1H NMR (600MHz, CDCl3),δH:9.17(br s,2H,O==C—NH—),8.69(s,1H,PhH),8.26~8.28 (d, 2H,J= 7.8 Hz, PhH), 7.58~7.60 (t, 1H,J=7.8Hz, PhH), 4.96 (br s, 4H, —O—CH2—CH2—N+—),4.81 (s, 4H, —N+—CH2—C==O), 4.43 (br s, 4H,—O—CH2—CH2—N+—),3.61[s,12H,—N+—(CH3)2],3.05~3.06 (br s, 4H, —NH—CH2—), 1.45~1.46 (br s,4H,—NH—CH2—CH2—),1.22~1.31[m,44H,—(CH2)11—],0.87~0.89[t,6H,J=7.2Hz,—(CH2)11—CH3]。
目 标 产 物I16:1H NMR (600MHz, CDCl3),δH:9.17(br s,2H,O==C—NH—),8.72(s,1H,PhH),8.28~8.30 (d, 2H,J=7.2Hz, PhH), 7.60~7.62 (t, 1H,J=7.2Hz, PhH), 4.96 (br s, 4H, —O—CH2—CH2—N+—),4.83 (s, 4H, —N+—CH2—C==O), 4.40 (s, 4H,—O—CH2—CH2—N+—), 3.60 [s, 12H, —N+—(CH3)2],3.04~3.07 (m, 4H, —NH—CH2—), 1.43~1.47 (m,4H,—NH—CH2—CH2—),1.22~1.31[m,52H,—(CH2)13—],0.87~0.89[t,6H,J=6.9Hz,—(CH2)13—CH3]。
根据1H NMR 数据可以看出,不同官能团中氢原子的化学位移与理论值相吻合,结合红外光谱和核磁氢谱数据分析,证明所合成化合物为目标产物。
2.2 电导率测试
在298.15K、308.15K、318.15K 和328.15K 条件下,分别测定了目标产物不同浓度水溶液的电导率值,绘制κ-C曲线,通过线性拟合得到两条直线的交点,交点所对应的浓度值即为CMC,如图6所示。
由图6可知,同一温度下,合成的目标产物溶液电导率随浓度升高而增大,在达到CMC 拐点之后,电导率增加幅度减缓,是由于当目标产物的浓度达到CMC 后,溶液中部分反离子(Cl-)被吸附在胶束表面,从而使胶束表面部分电荷被中和,使溶液中导电的自由移动离子减少,电导率增加的幅度减慢[22]。在同一浓度下,随着温度的升高,电导率增大,CMC 增加,是因为随着温度的升高,目标产物分子间的热运动加快,导致分子之间不容易形成胶束。胶束的离子化程度(α)由电导率曲线拐点后和前的两条线段的斜率比得到,而胶束的反离子结合度(β)[23]可按式(2)计算。


图6 不同温度下目标产物的电导率(κ)-浓度(C)曲线图
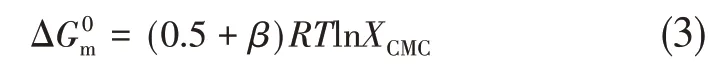
式中,XCMC是临界胶束浓度下表面活性剂与水的摩尔比,其值为CMC/55.4(25℃时,1L H2O 的摩尔数为55.4mol);T为热力学温度,K;R为理想气体常数。


不同温度条件下的CMC 以及胶束形成热力学参数如表1所示。

表1 目标产物的热力学参数和CMC
由表1 可知,在温度为298.15K 时,目标产物I12、I14和I16的CMC比结构相似的表面活性剂[16]CMC(1×10-4~1.17×10-3mol/L)降低了一个数量级。这主要是因为目标产物的分子结构中同时引入了酯键和酰胺键,使分子间更易形成氢键,更有利于促进分子间聚集,形成胶束,从而使CMC 更低,使目标产物具有更好的表面活性。

2.3 表面张力测试
在298.15K条件下,目标产物的γ-C曲线如图7所示,通过线性拟合得到曲线的拐点,拐点所对应的横坐标即为CMC,纵坐标即为γCMC。

图7 目标产物的表面张力(γ)-浓度(C)曲线
由图7 数值再结合式(6)~式(9)[26]可以计算出目标产物水溶液的表面饱和吸附量Γmax、目标产物分子在空气/水界面的最小横截面积Amin、效率因子pC20和CMC处的表面压πCMC,所有的表面张力参数均列于表2中。
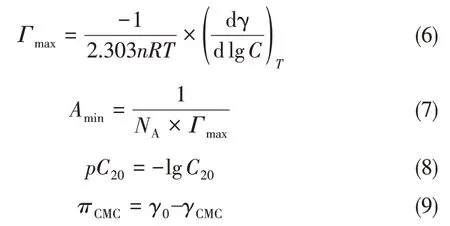
式中,R为 摩 尔 气 体 常 数,8.314J/(mol·K);T为热力学温度,K;NA为阿伏伽德罗常数,6.02×1023;γ为表面张力,mN/m;γ0为298K 下水的表面张力,71.25mN/m;n为常数,对于Gemini表面活性剂,取3[27]。
由表2 可知,随着疏水碳链长度的增加,Amin值增加,说明目标产物的疏水尾链在空气/水界面容易弯曲,从而导致在空气/水界面的聚集密度减小,Amin值增大;随着疏水碳链长的增加,pC20值增大,与胶束化过程相比,具有较长疏水碳链的I16更容易在空气/水界面吸附。目标产物与结构相似的表面活性剂[16]对比可知,相同疏水碳链长度的目标产物I16的γCMC比文献值降低了5.45mN/m,pC20更大,表明降低表面张力的效率更高。二者只是含官能团种类和数量不同,前者比后者多两个酯键,在气/液界面上的定向作用更强,使得疏水链伸到空气侧[28],有效降低了目标产物的表面张力。

表2 目标产物的表面参数(25℃)
2.4 泡沫性测试
测试目标产物和对比化合物[16]的起泡性和稳泡性,结果如图8和图9所示。

图8 目标产物和对比化合物的泡沫高度
由图8 和图9 可知,目标产物的起泡能力和稳泡能力均高于文献[16]。其中I8的起泡能力最好,初始泡沫体积最高可达40mL,I12和I14的稳泡性最好,均达到100%。这是由于目标产物分子结构中引入酯键,使得分子间氢键的相互作用加强,从而更有效地降低了水的表面张力,提高了界面膜的弹性和强度,因而起泡能力和稳泡能力优于对比文献[16]。
2.5 乳化性测试
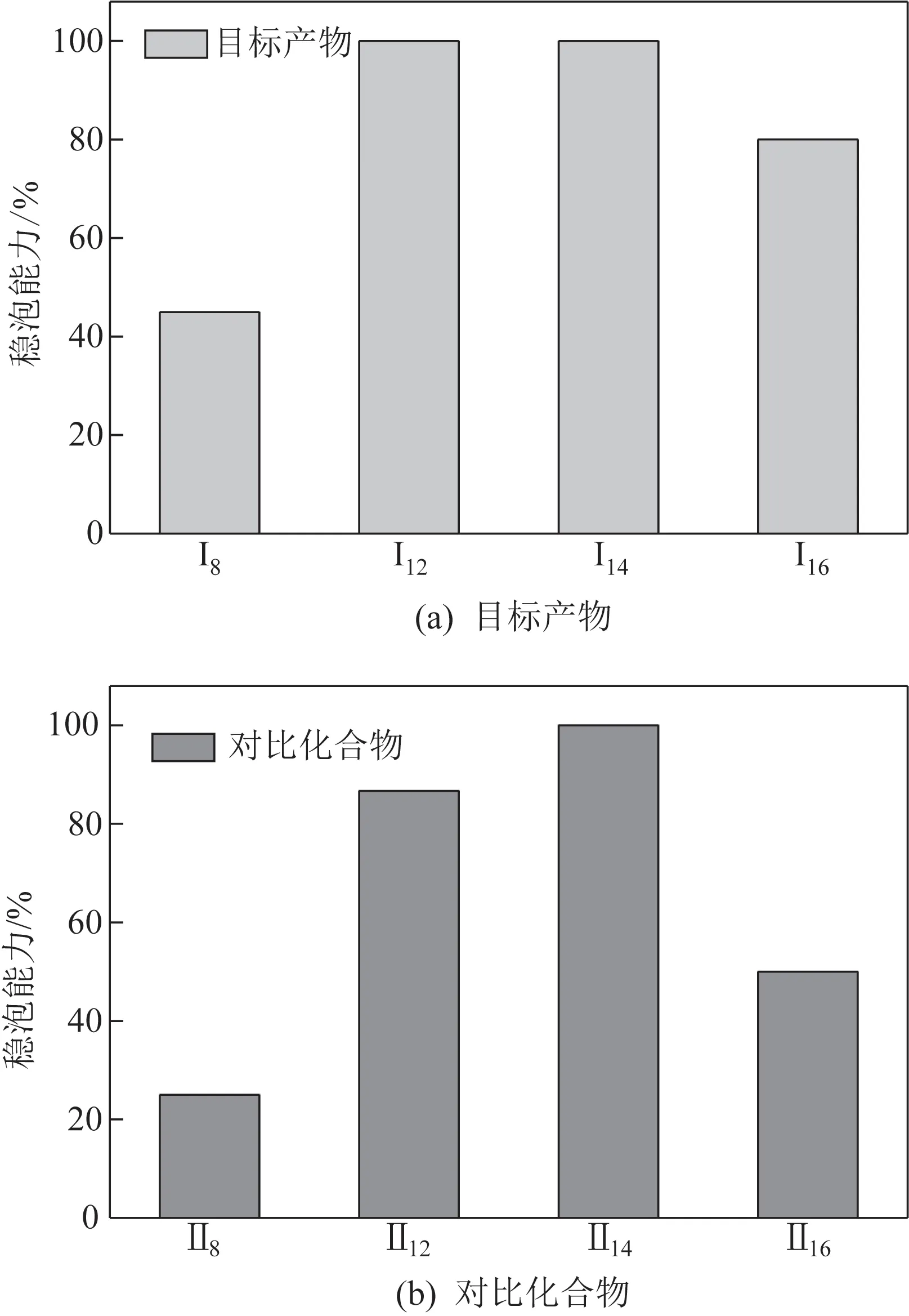
图9 目标产物和对比化合物的稳泡能力
测试目标产物和对比化合物[16]的乳化能力,乳化剂分出10mL 水所用的时间越长,表明乳化能力越好,结果如图10所示。
由图可知,目标产物对石蜡、机油、甲苯和二甲苯的乳化性结果表明,目标产物的乳化时间均高于对比化合物的乳化时间。这是由于目标产物分子结构中同时含有酯键和酰胺键,更容易产生分子间氢键和双电层结构,在降低表面张力的同时在界面吸附形成的界面膜更加坚韧不易破裂,有较强的稳定性,因此乳化能力较高,与文献阐述一致[29]。
由图11 可知,在对4 种物质的乳化性测试中,I12对二甲苯的乳化性最好,分出10mL 水的时间为900s,I16对苯的乳化性最好,分出10mL 水的时间为629s。
3 结论
(1)通过酰胺化和季铵化两步反应,合成系列目标产物I8、I12、I14和I16,通过FTIR、1H NMR 对结构进行表征,确定合成产物为目标产物。
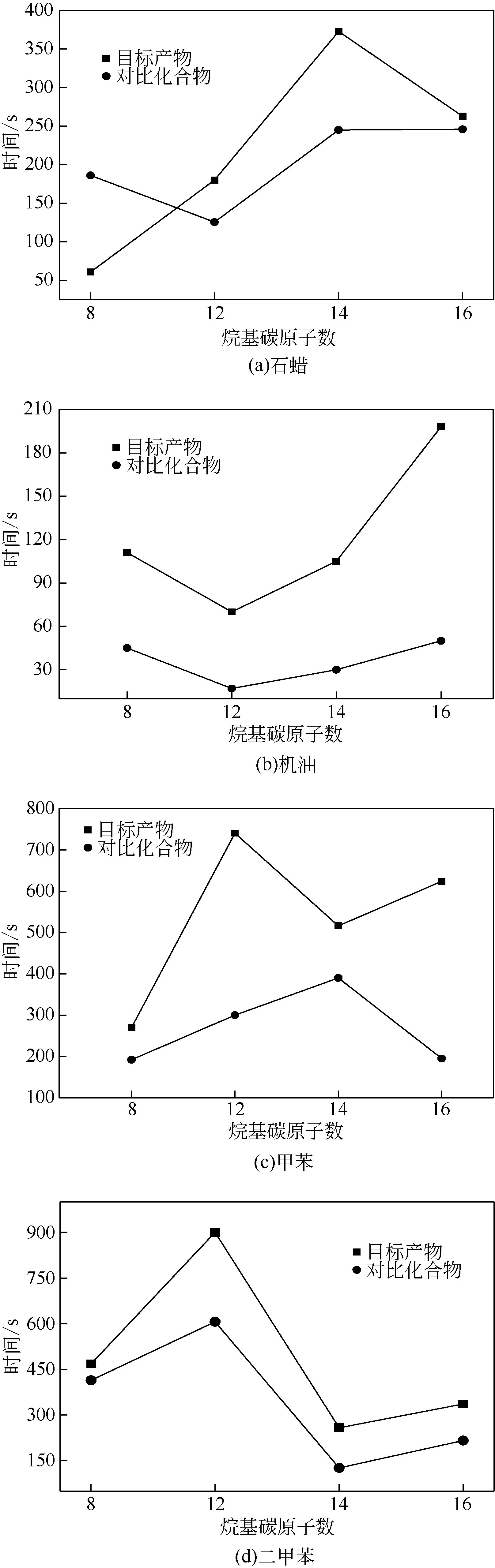
图10 目标产物和对比化合物的乳化能力

图11 目标产物的乳化能力
(3)泡沫性结果表明,与对比化合物相比,合成的目标产物具有很好的起泡性和稳泡能力,其中I8的起泡能力最好,初始泡沫体积达到40mL,I12和I14的稳泡性最好,均达到100%。
(4)乳化性能结果表明,与对比化合物相比,在合成的目标产物中I12对二甲苯的乳化性最好,分出10mL水的时间为900s;I16对苯的乳化性最好,分出10mL水的时间为629s。
本文所合成表面活性剂具有很好的表面活性,起泡能力、稳泡能力和乳化能力均较好,期望其在实际应用方面具有广阔的应用前景。
