Combining protein arginine methyltransferase inhibitor and anti-programmed death-ligand-1 inhibits pancreatic cancer progression
2020-08-17NanNanZhengMinZhouFangSunManXiuHuaiYiZhangChunYingQuFengShenLeiMingXu
Nan-Nan Zheng, Min Zhou, Fang Sun, Man-Xiu Huai, Yi Zhang, Chun-Ying Qu, Feng Shen, Lei-Ming Xu
Abstract
Key words: Protein arginine methyltransferase; Programmed death-ligand-1 blockade; Pancreatic ductal adenocarcinoma; Combination therapy; Tumor microenvironment
INTRODUCTION
Pancreatic ductal adenocarcinoma (PDAC) is the seventh leading cause of cancerrelated death worldwide and the fourth leading cause of cancer-related death in the United States[1,2]. Most cases of PDAC are diagnosed at the metastatic stage, and the median survival of these patients is less than one year[2]. Many factors are associated with the poor survival of PDAC patients and the treatment challenges, including the lack of early detection, high risk of relapse after curative surgery, and poor response to chemotherapy, radiation, molecular targeted therapy, and immunotherapy[3]. Programmed death-ligand-1 (PD-L1), also known as B7-H1, is a cell surface protein and one of two ligands for programmed death-1 (PD-1), a costimulatory molecule that negatively regulates T cell responses[4]. Ligation of PD-L1 on cancer cells to PD-1 on T cells suppresses T cell activation and proliferation. While immunotherapy using anti-PD-1/PD-L1 antibodies has been shown to be effective for many types of malignancies[5,6], its activity is limited in PDAC[7]. The mechanism underlying pancreatic cancer resistance to anti-PD-1/PD-L1 immunotherapy is poorly understood, but it has been suggested that PD-L1 expression in tumors is an important indicator of checkpoint immunotherapy efficacy[8,9].
Protein arginine methylation is a common posttranslational modification that plays a role in multiple pathways, including cell cycle control, RNA processing, and DNA replication[10]. Protein arginine methyltransferases (PRMTs) are enzymes that catalyze the transfer of a methyl group from S-adenosylmethionine to arginine[10]. PRMT family members are classified into PRMT types I, II, and III based on the nature of the catalyzed methylation reaction[11]. PRMT1 is the predominant type I methyltransferase responsible for approximately 85% of all cellular arginine methylation events. PRMT1 is deregulated in a wide variety of cancer types,e.g., pancreatic adenocarcinoma[12,13], gastric cancer[14], and lung cancer[15]. This enzyme controls epithelial-mesenchymal transition in cancer cells[16]. PRMT1 can catalyze arginine methylation on histones and other proteins, such as Axin and epidermal growth factor receptor[17,18]. PRMT1 is upregulated in pancreatic cancer and exerts an oncogenic role by regulating the βcatenin protein level[13].
The aim of the present study was to evaluate the effect of type I PRMT inhibitor against PDAC in mice and to investigate the influence of PRMT1 on PD-L1 expression.
MATERIALS AND METHODS
Cell culture
The murine PDAC cell line Panc02, which is syngeneic to C57BL/6 mice, was obtained from the cell bank of the Type Culture Collection of the Chinese Academy of Sciences (Shanghai, China). Panc02 cells were cultured in DMEM (Gibco, Grand Island, NY, United States) with 10% fetal bovine serum (Gibco) at 37°C in a 5% CO2atmosphere.
Mice and reagents
Female C57BL/6 mice (specific pathogen-free grade) aged 5 wk were obtained from Shanghai Jihui Experimental Animal Feeding Co., Ltd and housed in a specific pathogen-free facility (23°C, 12 h/12 h light/dark cycle, 50% humidity, and ad libitum access to food and water). All experimental procedures were approved by the ethics committee of Xinhua Hospital Affiliated to Shanghai Jiao Tong University School of Medicine, and the protocols adhered to approved institutional protocols set by the China Association of Laboratory Animal Care. Panc02 cells (5 × 106) suspended in 100 μL cold PBS were subcutaneously injected into the lower back region of each mouse. The tumor volume was calculated using the following formula: 0.52 × length × width2. When the tumors reached approximately 100 mm3, the tumor-bearing mice were randomly divided into four groups (n= 4) that were treated with control solvent (PBS, once daily), PT1001B (30 mg/kg, once daily, synthesized by Wanget al[19]), anti-PD-L1 mAb (200 μg/mouse, every 2 d for 5 intervals, Clone No. 10F.9G2, BioXcell), or PT1001B + anti-PD-L1 mAbviaintraperitoneal injection. Mice were sacrificed at 37 d following the initial injection, and tumors were removed and weighed.
Flow cytometry analysis
Immunophenotypic analyses of splenocytes and single-cell suspensions from tumors were assessed by flow cytometry (FCM). All primary antibodies used in this study were purchased from BioLegend (CA, United States). Cells were stained with antibodies specific for CD45-APC-Cy7 (Clone 30-F11), CD4-PE-Cy7 (Clone RM4-4), CD8-FITC (Clone 53-6.7), PD-L1-APC (Clone 10F.9G2), and PD-1-PE (Clone RMP1-30). For the apoptosis analysis, the cells were stained with annexin V-fluorescein isothiocyanate (FITC) and propidium iodide (PI) (BD apoptosis assay kit, BD Pharmingen, CA, United States) according to the manufacturer’s protocol. Fresh PDAC tumor tissues from the mouse model were minced into small pieces and then digested with collagenase type IV to generate a single-cell suspension. After being filtered and washed with cold PBS, the cells were incubated with the primary antibodies on ice for 30 min, washed, fixed in PBS containing 1% formalin, and analyzed on a flow cytometer (CyAn ADP, Beckman). Data were visualized using FlowJo software.
Immunohistochemistry
The tumor tissues isolated from sacrificed mice were immediately fixed in 4% paraformaldehyde for 24 h and embedded in paraffin. The embedded sections were sliced into 5-μm sections for staining. The deparaffinized and rehydrated sections were boiled in a high-pressure pot with sodium citrate antigen retrieval solution for 3 min. After three washes in PBS, the sections were incubated with 3% hydrogen peroxide in methanol for 15 min to inhibit endogenous peroxidase activity. After nonspecific reactions were blocked with 10% normal rabbit serum, the sections were incubated overnight at 4°C with rabbit polyclonal antibodies specific to Ki67 (GB13030-2, 1:200, Servicebio). Then, the sections were washed, incubated at room temperature for 50 min with horseradish peroxidase-conjugated secondary antibody (GB23303, 1:200, Servicebio), and counterstained with hematoxylin.
TUNEL assay
The TUNEL assay was performed according to the kit protocol (11684817910, Roche). Briefly, tumor tissue sections were deparaffinized in xylene, rehydrated in PBS, and incubated with proteinase K working solution for 25 min at 37°C. After three washes in PBS, the sections were incubated in permeabilization working solution for 20 min. Then, TdT and dUTP were mixed at a 1:9 ratio; the tissue samples were incubated with the resulting mixture in a flat wet box at 37°C for 3-4 h. After three washes in PBS, the slides were immersed in 3% H2O2at room temperature for 15 min in the dark. After three washes in PBS, the specimens were covered with converter-POD for 30 min and then washed three times in PBS. The slides were visualized with the DAB substrate and observed by microscopy (OLYMPUS, Japan). TUNEL-positive cells were counted with ImageJ, and the apoptotic index was calculated as the ratio of apoptotic cells to total cells in each field.
Immunofluorescence
After antigen retrieval, nonspecific binding was blocked with 1% bovine serum albumin for 30 min, and the tissue sections were incubated overnight at 4°C with a primary antibody against PD-L1 (GB11339, 1:200, Servicebio). Thereafter, the sections were incubated with Alexa Fluor®488-conjugated goat anti-mouse IgG (GB25301, 1:400, Servicebio) for 50 min at room temperature. After incubation with CY3 reagent for 10 min, the sections were heated in a microwave to remove antibodies bound to the tissue. After nonspecific binding was blocked, the samples were incubated overnight at 4°C with a primary antibody against PRMT1 (sc-166963, 1:1000, Santa Cruz). Thereafter, the sections were incubated with horseradish peroxidase-labeled goat antimouse IgG (GB23301, 1:500, Servicebio) secondary antibody for 50 min at room temperature. Nuclei were stained with DAPI for 5 min at room temperature. Immunopositive cells were analyzed using a fluorescence microscope (Eclipse ci, NIKON, Japan).
Statistical analysis
All data are shown as the mean ± SE. All data were assessed byt-tests or one-way ANOVA (GraphPad Prism 6.0).Pvalues < 0.05 were considered to indicate statistical significance.
RESULTS
PD-L1 is expressed in PDAC in vivo but not in vitro
To determine PD-L1 expression in tumor-bearing mice, we transplanted Panc02 cells into C57BL/6 mice (Figure 1A). PD-L1 expression on tumor cells was quantified by the mean fluorescence intensity (MFI) (Figure 1B). Panc02 cells grownin vitrodid not express PD-L1, but subcutaneous Panc02-derived tumor cells expressed PD-L1. We speculated that the increased PD-L1 expressionin vivois related to the tumor microenvironment.
PT1001B reverses anti-PD-L1 resistance in a PDAC mouse model
We then used PT1001B (formerly known as compound 28d, DCPR049_12), a novel selective inhibitor of type I PRMTs that effectively inhibits cancer cell proliferation[19]. We wondered whether PT1001B can enhance the efficacy of anti-PD-L1 therapy in Panc02-bearing C57BL/6 mice. The tumor-bearing mice were treated with PT1001B and anti-PD-L1 mAb, alone or in combination. As shown in Figure 2A, the tumors were resistant to anti-PD-L1 monotherapy. PT1001B or anti-PD-L1 mAb therapy alone did not decrease tumor growth compared to no treatment. Interestingly, when the anti-PD-L1 mAb was combined with PT1001B, the tumors showed a better response, as assessed by tumor volume (1054.00 ± 61.37 mm3vs555.80 ± 74.42 mm3) and weight (0.83 ± 0.06 gvs0.38 ± 0.02 g) (Figure 2B). Importantly, treatment with PT1001B and PD-L1 blockade, alone or in combination, did not result in any overt signs of toxicity, as evidenced by weight gain in all the evaluated animal groups (Figure 2C). There was no difference in mouse body weight in different treatment groups. Therefore, these findings suggest the potential of combination therapy with PT1001B to reverse anti-PD-L1 resistance in PDAC.
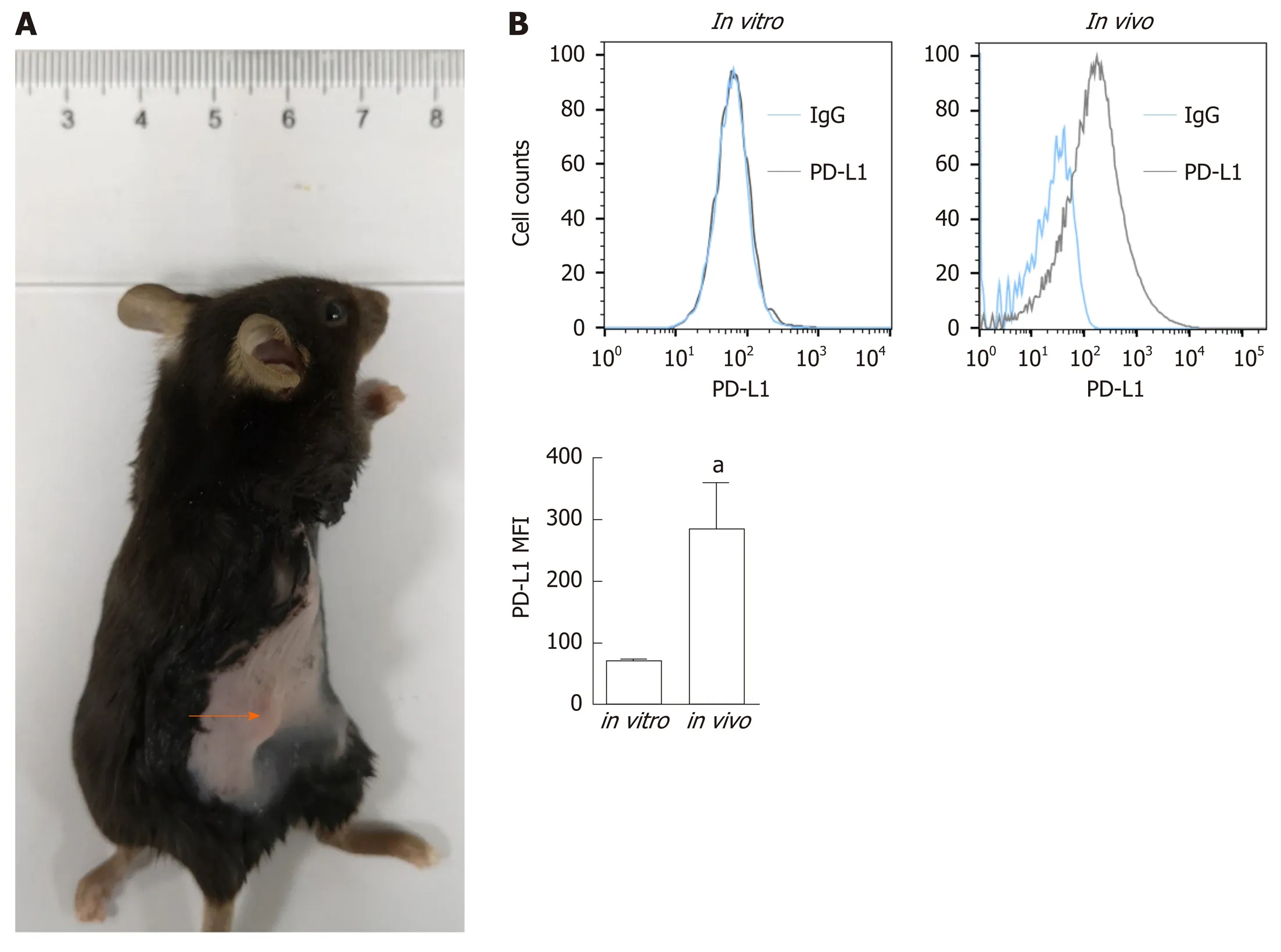
Figure 1 Programmed death-ligand-1 expression in pancreatic ductal adenocarcinoma in vitro and in vivo. A: Panc02 cells were subcutaneously transplanted into C57BL/6 mice to establish pancreatic tumors (orange arrow); B: Programmed death-ligand-1 expression levels in cultured pancreatic cancer cells (in vitro) and tumors (in vivo). The mean fluorescence intensity was used to quantify protein expression levels, and the values were statistically analyzed by a two-sided t-test; the results are presented at the bottom. Data represent the mean ± SE (n = 3). aP < 0.05 vs in vitro condition. PD-L1: Programmed death-ligand-1; MFI: Mean fluorescence intensity.
PT1001B enhances antitumor immunity
To determine whether PT1001B can enhance antitumor immunity, single-cell suspensions of tumor and spleen extracts were stained for FCM analysis. As shown in Figure 3A, PT1001B decreased PD-L1 expression on cancer cells (32.74% ± 5.89% in the control groupvs17.95% ± 1.92% in the PT1001B group). PD-L1 expression was dramatically lower in the anti-PD-L1 + PT1001B group than in the control group (4.21% ± 0.82%vs32.74% ± 5.89%). In tumors, PD-1+ leukocytes were downregulated in the three treated groups (19.93% ± 3.65% in the PT1001B group, 15.53% ± 1.71% in the anti-PD-L1 group, and 6.48% ± 1.08 % in the anti-PD-L1 + PT1001B groupvs35.77% ± 3.30% in the control group) (Figure 3B). In the spleen, PD-1+ leukocytes were downregulated to a greater extent in the anti-PD-L1 + PT1001B group than in the anti-PD-L1 group (5.98% ± 1.26% in the anti-PD-L1 + PT1001B groupvs10.35% ± 0.46% in the anti-PD-L1 group) (Figure 3C). Analysis of T cell populations from the tumors and spleens revealed that PT1001B further increased the anti-PD-L1 mAb-induced tumor infiltration of CD8+ cytotoxic T lymphocytes (CTLs) (tumor: 8.14% ± 0.82% in the anti-PD-L1 groupvs13.83% ± 0.97% in the anti-PD-L1+PT1001B group; spleen: 7.54% ± 1.09% in the anti-PD-L1 groupvs12.90% ± 0.15% in the anti-PD-L1 + PT1001B group) (Figure 3D and E). These observations suggest that PT1001B enhanced antitumor immunity by upregulating tumor infiltrating CD8+ T lymphocytes, decreasing PD-L1 expression by cancer cells, and downregulating PD-1+ leukocytes.
Effects of PT1001B and anti-PD-L1 mAb on PDAC cell proliferation and apoptosis
To estimate the effect of PT1001B on apoptosis, FCM analysis was performed after double staining with annexin V-FITC/PI. As shown in Figure 4A, the rates of early and late apoptosis were dramatically higher in the combination group than in the control group (49.00% ± 0.64%vs24.36% ± 3.67%), while the rates showed no significant changes in the other treatment groups compared to the control. We performed immunohistochemistry analysis of tumor tissues to examine Ki67 expression and TUNEL positive cells, which are common markers of proliferation and apoptosis, respectively. Consistent with the greater degree of tumor growth inhibition, PT1001B significantly increased the efficacy of anti-PD-L1 therapy in suppressing tumor cell proliferation and inducing tumor cell apoptosisin vivo(Figure 4B and C).

Figure 2 The combination of PT1001B and programmed death-ligand-1 blockade inhibits the development of pancreatic ductal adenocarcinoma in mice. Panc02 cells were surgically transplanted into C57BL/6 mice. One week later, tumors reached approximately 100 mm3, and tumorbearing mice were randomly divided into four groups that were treated with PT1001B (30 mg/kg body weight, once daily) or anti- programmed death-ligand-1 (PD-L1) mAb (200 μg/mouse, every 2 d for 5 intervals) alone or in combination via intraperitoneal injection for 37 d. A: The combination of PT1001B and anti-PD-L1 mAb significantly inhibited tumor growth; B: Tumors in different treated groups; C: Mouse body weight curve. The values were analyzed by one-way ANOVA. Data represent the mean ± SE (n = 4). aP < 0.05, bP < 0.01 vs control group. PD-L1: Programmed death-ligand-1.
PRMT1 expression correlates with PD-L1 expression
Immunofluorescence was performed to detect the levels of PRMT1 and PD-L1 in tumor tissues (Figure 5). PT1001B promoted the downregulation of PRMT1 and PDL1, as indicated by the reduction in fluorescence. In particular, the anti-PD-L1 + PT1001B group showed the most drastic effect, with the lowest intensity of PD-L1-positive staining in tumor tissue.


Figure 3 PT1001B enhances antitumor immunity. Cell suspensions were prepared from pancreatic tumor tissues and spleens from the four groups of tumor-bearing mice and stained with CD45-, CD4-, CD8-, PD-L1-, and PD-1-specific antibodies. Stained cells were analyzed by flow cytometry. The left panel presents representative images, and the right panel shows the statistical analysis. A: Analysis of CD45-/PD-L1+ cells in the tumors; B: Analysis of CD45+/PD-1+ cells in the tumors; C: Analysis of CD45+/PD-1+ cells in the spleens; D: Analysis of CD4+ T cells and CD8+ T cells in the tumors; E: Analysis of CD4+ T cells and CD8+ T cells in the spleens. Differences in the percentages of cells were analyzed by one-way ANOVA. Data represent the mean ± SE (n = 4). aP < 0.05, bP < 0.01 vs control group; cP < 0.05, dP < 0.01 vs anti-PD-L1 group. PD-L1: Programmed death-ligand-1; PD-1: Programmed death-1.
DISCUSSION
Immunotherapy failure in pancreatic cancer stems from the nonimmunogenic characteristic, especially the immunosuppressive microenvironment, poor T cell infiltration, and the low mutational burden, which contribute to creating an immunoprivileged environment[20]. Increased PD-L1 expression by cancer or stroma cells is a fundamental mechanism of escape from host immunity[21]. Tumor cells can inhibit the proliferation, survival, and effector function of T lymphocytes, especially CD8+ T lymphocytes, through the PD-L1/PD-1 pathway[22]. PD-L1 blockade can relieve immunosuppression, enhance antitumor immunity, and lead to the expansion of tumor-infiltrating lymphocytes[23]. Immune checkpoint blockade reverses immunosuppression to activate tumor-reactive CTLs that directly target tumor cells for apoptosis[24]. However, pancreatic cancer cannot be cured by PD-1/PD-L1 blockade alone because of the specific tumor microenvironment[25]. Therefore, the search for effective combination therapy will provide new clues that affect the efficacy of immunotherapy. It was previously reported that MEK inhibitors, BET inhibitors, and mTOR inhibitors decreased PD-L1 expressionin vitroandin vivo[26-28]. In our study, we identified a novel small-molecule inhibitor of type I PRMTs that also suppressed PDL1 expression in pancreatic cancer cells.
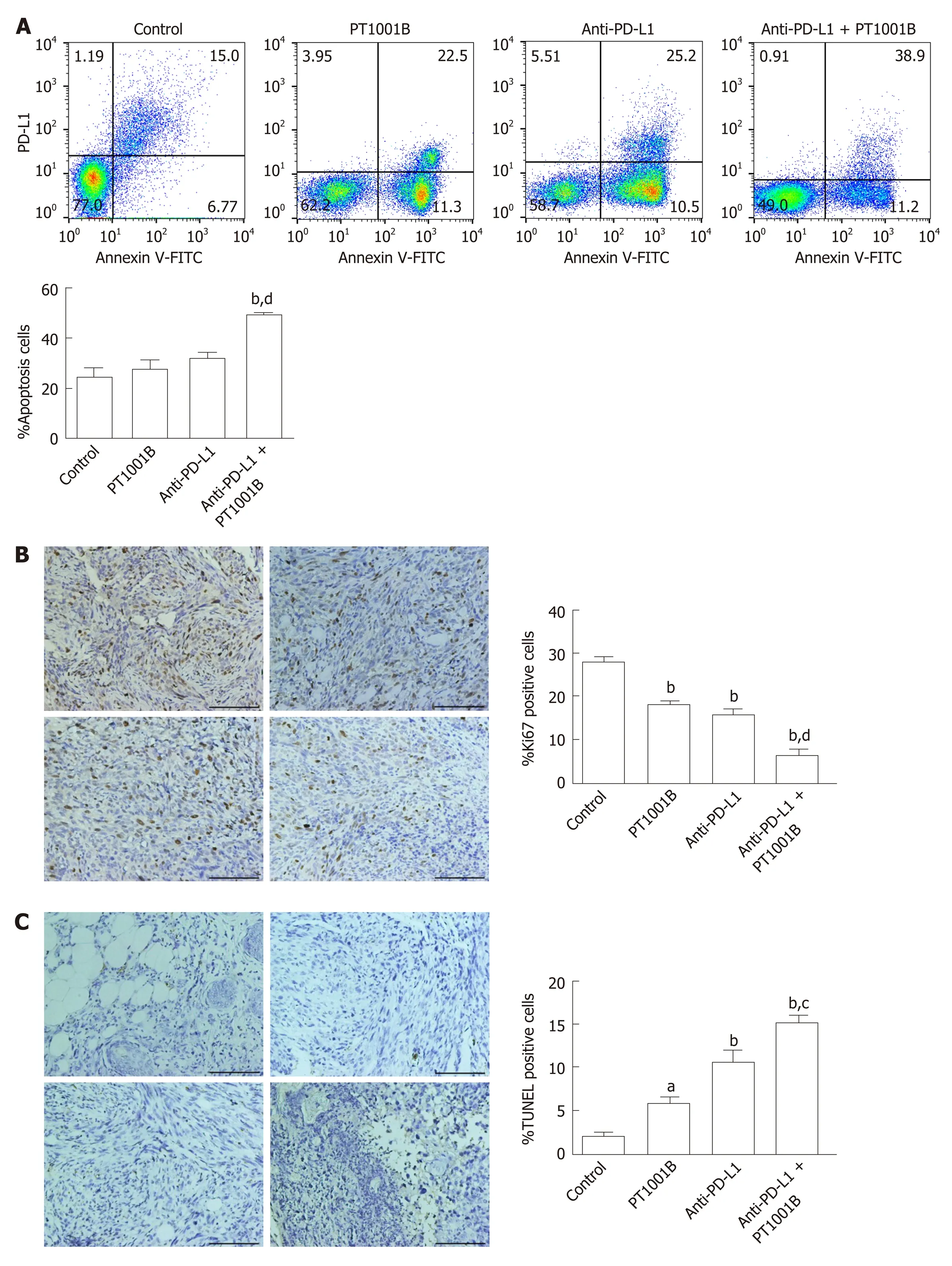
Figure 4 Effect of PT1001B and anti-programmed death-ligand-1 mAb on tumor cell proliferation and apoptosis. A: Cell suspensions prepared from pancreatic tumor tissues were stained with annexin V and propidium iodide to analyze the apoptosis rate by flow cytometry; B: The four groups of tumors were sectioned and stained for Ki67 (brown); C: The four groups of tumors were sectioned and subjected to TUNEL assay (brown). Representative images are shown. a: Control; b: PT1001B; c: Anti-programmed death-ligand-1 (PD-L1) mAb; d: Anti-PD-L1 mAb + PT1001B. Scale bars = 100 µm. The populations of apoptotic cells, Ki67-positive, or TUNEL-positive cells were analyzed by one-way ANOVA. Data represent the mean ± SE (n = 4). aP < 0.05, bP < 0.01 vs control group; cP < 0.05, dP < 0.01 vs anti-PD-L1 group. PI: Propidium iodide; FITC: Fluorescein Isothiocyanate; PD-L1: Programmed death-ligand-1; TUNEL: Terminal deoxynucleotidyl transferase mediated dUTP nick-end labeling.

Figure 5 Immunofluorescence analysis of protein arginine methyltransferase and programmed death-ligand-1 in tumor tissue. Protein arginine methyltransferase 1 is shown in red, programmed death-ligand-1 is shown in green, and DAPI staining indicates the nucleus. Scale bars = 100 µm. PRMT: Protein arginine methyltransferase; PD-L1: Programmed death-ligand-1; DAPI: 2-(4-Amidinophenyl)-6-indolecarbamidine dihydrochloride.
In the present study, we demonstrated that the combination of PD-L1 blockade and PT1001B showed increased efficacy. A limited response to anti-PD-L1 monotherapy was observed in our PDAC model. Interestingly, the resistance to anti-PD-L1 therapy was reversed by the addition of PT1001B. When analyzing the expression of key immune checkpoint proteins and immune infiltration in the tumor microenvironment and spleen, we observed that the proportion of tumor-infiltrating effector cells was increased and the proportion of PD-1+ leukocytes was decreased in the combination group. This result is significant, as limited T cell infiltration is considered a barrier to the efficacy of immunotherapy in PDAC. CTLs primarily use perforin and Fasmediated effector mechanisms to induce tumor cell apoptosis[29]. PD-1 is mainly expressed on activated CD4+ T cells, CD8+ T cells, and B cells in the periphery[30]. PD-1 inhibition can result in the loss of peripheral tolerance and an increase in autoimmunity[31]. In addition, PT1001B amplified the inhibitory effect of anti-PD-L1 on tumor cell proliferation and enhanced the induction of tumor cell apoptosis.
PRMT1 is the major methyltransferase in mammalian cells and is usually considered an epigenetic molecular marker. A previous study suggested that PRMT1 is overexpressed in pancreatic cancer cells and that elevated PRMT1 levels predict a poor clinical outcome. Moreover, PRMT1 knockdown was shown to inhibit tumor growthin vivo[13]. However, our results indicated that PT1001B alone did not decrease tumor growth. The discrepancy between our study and previous reports might be due to the use of different mouse models, cell lines, and inhibition methods. Our study results showed that PRMT1 downregulation was correlated with PD-L1 downregulation in PDAC tumors. Studies suggest that PRMT1 regulatesc-mycgene expression and function[32,33]. Current data show thatc-myc, an important signaling hub and driver gene, is commonly overexpressed and aberrantly activated in PDAC[34,35]. The activation ofc-mychas been shown to stimulate PD-L1 expression in some cancer cells, causing immune evasion[36], andc-mycregulates PD-L1 expression in PDAC[37]. Therefore, we speculated that PT1001B might downregulate PD-L1 expression by inhibiting the master transcription amplifierc-myc. One limitation of this study is that PT1001B inhibits four PRMTs in addition to PRMT1 (PRMT3, PRMT4, PRMT6, and PRMT8), suggesting that it is a pan inhibitor of type I PRMTs[19]. Therefore, the specificity of PT1001B as an epigenetic agent in cancer therapy remains to be further studied.
In summary, our data demonstrate that the combination of the type I PRMT inhibitor and PD-L1 blockade may be an effective therapeutic approach for PDAC. PRMT1 expression is correlated with PD-L1 expression, and PRMT1 is a potential therapeutic target. Additional research aimed at elucidating the mechanisms by which PRMT1 regulates PD-L1 protein levels is warranted.
ARTICLE HIGHLIGHTS

ACKNOWLEDGEMENTS
The authors would like to acknowledge Cheng Luo and Yuanyuan Zhang (Chinese Academy of Sciences Shanghai Institute of Materia Medica, Shanghai, China) for providing PT1001B.
杂志排行

World Journal of Gastroenterology的其它文章
- Nomogram for predicting transmural bowel infarction in patients with acute superior mesenteric venous thrombosis
- Functional gastrointestinal disorders in inflammatory bowel disease: Time for a paradigm shift?
- Intratumoral heterogeneity of hepatocellular carcinoma: From single-cell to population-based studies
- Helicobacter pylori-induced inflammation masks the underlying presence of low-grade dysplasia on gastric lesions
- Adipose-derived mesenchymal stem cells alleviate TNBS-induced colitis in rats by influencing intestinal epithelial cell regeneration, Wnt signaling, and T cell immunity
- Expression of Notch pathway components (Numb, Itch, and Siah-1) in colorectal tumors: A clinicopathological study