CaCO3 改性对提高MgO 基CO2 吸附剂循环稳定性的影响
2020-08-07柳春杰吴素芳
柳春杰, 吴素芳
(浙江大学 化学工程与生物工程学院, 浙江 杭州 310027)
1 前 言
近年来,以全球气候变化为核心的环境问题日益严重,而消减CO2等温室气体排放是当前减缓气候变化的重要途径之一。燃煤电厂烟气是CO2气体的主要排放来源。捕集这部分CO2是控制CO2排放的重要手段。燃烧前捕集技术主要应用于整体煤气化联合循环 (integrated gasification combined cycle 简称IGCC) 发电系统中[1-3]。因其捕集设备小、工艺成本低等优势成为未来火电厂改革发展的主线。
燃烧前捕集二氧化碳的方法有化学吸收法、吸附法、膜分离法和低温分离法等。其中,吸附法具有操作条件温和、能耗低、可再生能力强、吸附速率快等优点,成为当前最具有发展前景的一种捕集 CO2的方法。在吸附法中,吸附剂的选择至关重要,直接决定了CO2的捕集效果。由于IGCC 中燃烧前捕集CO2的合成气温度范围为250 ~ 400 ℃[4],所以应采用中温吸附剂。其中,MgO 基吸附剂具有较高的理论吸附容量,约为1.1 g·g-1,被认为是理想的中温CO2吸附剂。但是在实际应用中,MgO 基吸附剂的吸附速率较慢,实际吸附容量较低,一般低于0.01 g·g-1,且循环稳定性较差,这些问题都限制了MgO 基吸附剂在工业上的广泛应用[5]。为了提高 MgO 基吸附剂的吸附容量,研究人员采用多孔氧化铝、活性炭、介孔硅为载体的负载法[6-9],使吸附容量最大提高到0.085 g·g-1。
STEVEN 等[10]发表专利报道了一种新型的双盐修饰的MgO 基吸附剂,通过负载碱金属碳酸盐复盐,MgO 基吸附剂吸附容量最高可达0.53 g·g-1。后续的研究发现[11-13],在提高MgO 吸附容量方面,硝酸盐和碳酸盐起协同作用。此外,TAKUYA 等[14]湿混、煅烧法制备 (Li-Na-K)NO3修饰的MgO 基吸附剂,在300 ℃、100% CO2气氛下初次吸附容量为0.275 g·g-1;ANH 等[15]采用溶胶-凝胶法和超临界干燥法制备Na2CO3和 NaNO3修饰的 MgO 基吸附剂,在 325 ℃、100% CO2气氛下初次吸附容量为 0.31 g·g-1,14 次循环中吸附容量基本保持不变。王磊[16]采用沉淀-沉积法制备复合碱金属硝酸盐和碳酸盐修饰的MgO 基吸附剂,在350 ℃、40% CO2下初次吸附容量为0.67 g·g-1,12 次循环后吸附容量下降为0.52 g·g-1。分析认为,碱金属盐在高温下 (350 ~ 400 ℃) 呈熔融状态且容易大范围流动,导致吸附剂的循环稳定性较差。所以本文采用塔曼温度(又称泰曼温度,指的是固体晶格开始明显流动的温度,一般在固体熔点的2/3处的温度)高于碳酸镁的CaCO3作为惰性添加剂,使其可以在一定程度上起到阻隔作用,限制碱金属盐的流动;而且CaCO3呈碱性,可以促进MgO 基吸附剂的CO2吸附性能。
本文分别以碱式碳酸镁和柠檬酸镁作为 MgO 的前驱体、采用尿素水解沉积法制备 CaCO3、通过混合浸渍等方法制备一系列经 CaCO3改性的、碱金属盐修饰的 MgO 基吸附剂,研究 MgO 颗粒大小以及CaCO3添加量对MgO 基吸附剂的循环稳定性的作用机制。
2 实验部分
2.1 试剂
碱式碳酸镁、轻质氧化镁、一水合柠檬酸、四水合硝酸钙、尿素、硝酸钾、硝酸锂、无水碳酸钾、和无水碳酸钠,以上化学试剂均为分析纯。无水甲醇,99.5% (质量分数)。以上化学试剂均采购自国药集团化学试剂有限公司。
2.2 吸附剂的制备
吸附剂制备流程如图1 所示。先进行MgO 前驱体的制备,CaCO3的制备并与MgO 前驱体混合,煅烧后再浸渍碱金属盐。
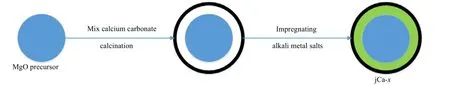
图1 吸附剂制备流程示意图Fig.1 Schematic diagram of the preparation process of adsorbents
2.2.1 MgO 前驱体的制备
将轻质MgO 加入去离子水中制成悬浮液,将配制好的柠檬酸溶液逐滴加入悬浮液中,50 ℃下水浴加热2 h 形成溶胶,升温至80 ℃继续加热蒸干水分,放入烘箱中110 ℃下烘8 h,制备得到柠檬酸镁前驱体。碱式碳酸镁不需另外制备,采用商业碱式碳酸镁。
2.2.2 CaCO3的制备与添加
将摩尔比为1:4 的硝酸钙与尿素溶于去离子水中,搅拌使其充分溶解;将MgO 前驱体 (碱式碳酸镁或者柠檬酸镁) 加入到上述溶液中超声分散10 min,将此混合液于80 ℃下搅拌,过滤洗涤,在60 ℃下烘干5~8 h;随后在马弗炉中500 ℃下煅烧2 h。
2.2.3 碱金属盐的浸渍
将硝酸锂、硝酸钾、碳酸钠、碳酸钾 (总碱金属盐与MgO 的摩尔比为0.15,硝酸盐与碳酸盐的摩尔比为2;其中硝酸锂与硝酸钾摩尔比为0.44 ∶ 0.56,碳酸钠与碳酸钾摩尔比为0.5:0.5) 溶解于无水甲醇中,超声分散使其完全溶解;称取0.4 ~ 0.5 g 上述煅烧后的固体加入溶液中,使其充分混合;水浴加热将无水甲醇蒸发;蒸干后,将白色固体置于烘箱中,80 ℃下烘干,称量所得样品并记录。
未添加CaCO3的吸附剂分别命名为jCa-0 和nCa-0,j 表示碱式碳酸镁为前驱体、n 表示柠檬酸镁为前驱体。将添加CaCO3的质量含量分别为3%、5%、10% 和20%的样品分别命名为jCa-1、jCa-2、jCa-3和jCa-4。
2.3 吸附剂的表征
通过X 射线衍射仪 (x-ray diffraction, XRD, D/MAX-RA, Rigaku, Japan) 10~80°扫描来表征吸附剂的晶形组成;采用日本HITACHI 公司生产的S-4800 型扫描电子显微镜 (scanning electron microscope, SEM)观察吸附剂的表观形貌、颗粒的大小等;采用日本BEL 公司生产的SORP-mini II 型测试仪,在液氮环境下检测CO2吸附剂的孔结构,采用多层吸附模型BET (Brunauer-Emmer-Teller) 公式计算样品的比表面积,BJH (Barrett-Joyner-Halenda) 公式分析样品的孔容和孔径分布。
2.4 CO2 吸附性能的测试
采用美国Perkin Elmer 公司生产的Pyris1 热重分析仪 (thermo gravimetric analyzer,TGA) 对制备的吸附剂进行吸附性能的测试。测试条件: 1.5 ~2.0 mg 的样品,首先在N2气氛下升高到400 ℃保持30 min,除去吸附剂中的水分和其他杂质;吸附时温度为350 ℃,吸附时长为60 min,气体氛围为40% CO2与60%N2混合气;脱附时温度为400 ℃,脱附时长为20 min,气体氛围为100% N2;吸脱附反复切换实现吸附剂的循环性能测试。
根据 TGA 实时记录的重量变化的数据,可以计算出单次吸附中吸附剂的吸附容量和吸附速率;通过比较吸附容量随着循环次数增加的变化可以用来表征吸附剂的吸附稳定性好坏。吸附容量的计算公式如下:
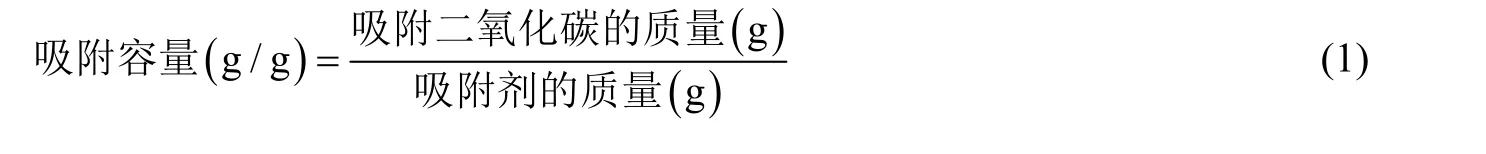
3 结果与讨论
3.1 前驱体种类对MgO 颗粒大小的影响
将两种前驱体煅烧后的样品进行XRD 射线分析,结果如图2 所示。可以发现,碱式碳酸镁和柠檬酸镁煅烧后均主要生成MgO,分别记为MgO(1)与MgO(2)。采用如下所示的谢乐公式计算晶粒尺寸。
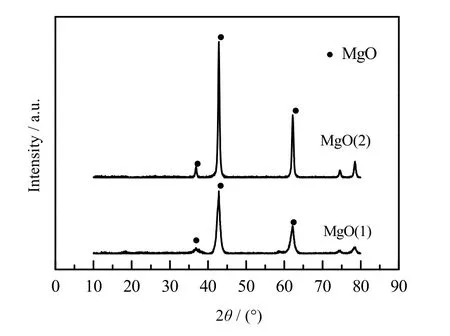
图2 前驱体煅烧后的MgO 的XRD 谱图Fig.2 XRD spectra of MgO after calcination of precursors

计算可知,MgO(1)的晶粒尺寸范围为 8~12 nm,MgO(2)的晶粒尺寸范围为18~22 nm。由此可知,碱式碳酸镁作为前驱体煅烧得到的MgO 颗粒更小。
采用热重分析仪对碱式碳酸镁和柠檬酸镁的分解温度进行测定,如图3 所示。进行对比发现,碱式碳酸镁在450 ℃下分解最快,到500 ℃左右分解完全;而柠檬酸镁在550 ℃左右分解最快,到600 ℃分解完全。可见,柠檬酸镁的分解温度更高,高温下MgO 颗粒更易于发生烧结团聚,所以柠檬酸镁煅烧制得的 MgO颗粒更大。
将两种前驱体煅烧浸渍后制备的吸附剂进行CO2吸附循环稳定性的测试,结果如图4 所示。由图可见,碱式碳酸镁制备的 jCa-0 吸附容量较高,但吸附稳定性较差;而柠檬酸镁制备的nCa-0 吸附容量较低,但循环稳定性较好。表明前驱体的种类对吸附剂的吸附性能有一定影响。分析原因可知,jCa-0 中MgO 颗粒较小,其活性组分分散性较好,所以初次吸附容量较高,为0.51 g·g-1。而nCa-0 中MgO 颗粒较大,初次吸附容量就相对较低,为0.20 g·g-1。其次,随着循环次数的增加,两种吸附剂的CO2吸附容量均有一定的降低。经20 次循环后,jCa-0吸附剂吸附容量从0.51 下降到0.25 g·g-1,而nCa-0 吸附剂的吸附容量从0.20 下降到0.13 g·g-1。吸附容量下降的原因可能是,在循环反应的过程中,MgO 颗粒逐渐团聚成大颗粒,且熔融状态的碱金属盐发生流动导致其分布不均匀,使其促进CO2吸附的效果有所降低。综合比较两者的吸附性能,虽然nCa-0 在循环过程中衰减率较低,但其初始吸附容量太低,所以本文后续采用碱式碳酸镁做进一步研究。
3.2 CaCO3 含量对循环稳定性的影响
3.2.1 吸附剂的表征
图5 为jCa-0 和jCa-2 (CaCO3的质量分数为5%) 浸渍碱金属盐前后的SEM 图。由图5(a)可见, MgO(1)(碱式碳酸镁煅烧后得到) 呈颗粒状。由图 5(c) 可见,掺杂 CaCO3改性制备的吸附剂呈片状,且改性后的吸附剂颗粒间空隙较大;这表明,CaCO3的添加,提高了活性组分MgO 颗粒的分散性。对比图5(a)、(b)两图可以看出,浸渍碱金属盐后,MgO 颗粒间的空隙被填充;但从图 5(d)可以看出,掺杂 CaCO3改性的吸附剂颗粒间仍有较大空隙,这也有助于CO2气体的吸附、脱附,一定程度上可以提高吸附剂的吸附速率。
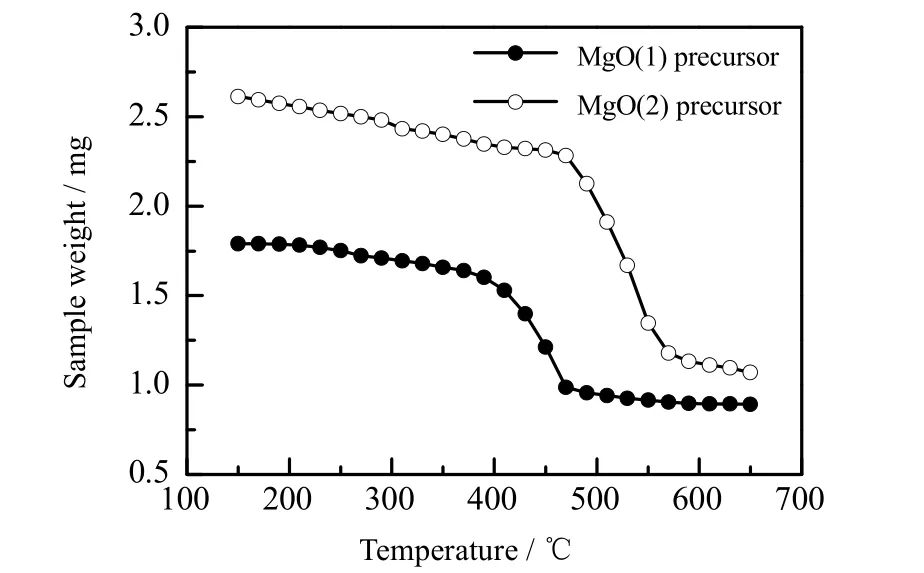
图3 MgO 前驱体分解温度曲线Fig.3 Decomposition temperature curves of MgO precursors

图4 吸附剂的循环吸附容量与循环次数的关系图Fig.4 Relationship between adsorption capacity and cycle numbers

图5 样品的SEM 图Fig.5 SEM images of samples
图6 为jCa-2 浸渍碱金属盐前后的XRD 图。从图中可以观察到CaCO3的衍射峰,说明尿素水解法成功地将CaCO3添加到了 MgO 基吸附剂中。图中也可以观察到KNO3、Na2CO3和 K2CO3的衍射峰,但是并未发现 LiNO3的衍射峰,分析其原因可能是 LiNO3在 MgO 表面上高度分散、且含量比较少,所以其衍射峰比较弱,很难检测出来。
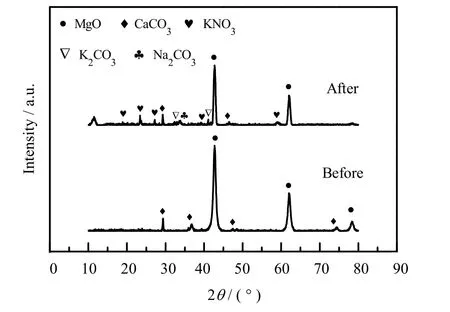
图6 jCa-2 浸渍前后的XRD 谱图Fig.6 XRD spectra of jCa -2 before and after impregnation
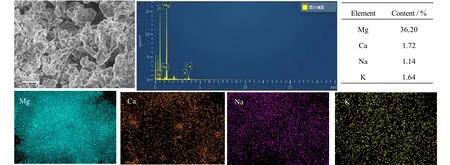
图7 jCa-2 的 EDS 以及元素含量图Fig.7 EDS images and element contents of the jCa-2 adsorbent
图7 为jCa-2 的能谱分析(EDS)以及元素含量图。从图中可以看出,Ca 的分布较均匀,且能谱显示Ca 元素的质量分数为1.72%,Mg 元素为 36.20%,换算得到CaCO3在吸附剂中的质量分数为4.5%,与添加值5%较为接近,说明尿素水解法可以很好的将CaCO3与MgO 混合。碱金属盐通过浸渍法添加到吸附剂表面,图7 显示Na、K 的分布也较为均匀,通过计算发现实际含量与添加含量相近,说明浸渍法中元素损失较少。但是由于EDS 的测试原理限制,Be 元素之前的元素不能被检测出来,所以添加的Li 未能在EDS 中显示分布和含量。
图8 表示不同CaCO3含量的吸附剂孔径分布图。可以看出,jCa-1、jCa-2 和jCa-3 三个样品的孔径分布基本相同,但是 jCa-2 和jCa-3 改性吸附剂的孔体积比jCa-1 改性吸附剂的大,故有利于CO2的扩散,并可以提高CO2的吸附速率;而jCa-1 改性吸附剂在0 ~10 nm 的孔数量较少,所以CO2扩散较慢,导致其诱导期 (从通入CO2气体开始到发生反应的时间段) 较长。 表1 为不同CaCO3含量吸附剂的比表面积值。由表可知,CaCO3含量对吸附剂的比表面积影响不大。
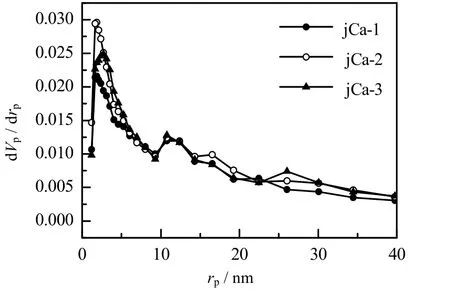
图8 不同CaCO3 含量的吸附剂的孔径分布图Fig.8 Pore size distribution of adsorbents with different CaCO3 contents

表1 不同碳酸钙含量吸附剂的比表面积Table 1 Specific surface areas of adsorbents with different CaCO3 contents
3.2.2 吸附容量和吸附速率测定
添加 CaCO3改性的吸附剂,其吸附容量随 CaCO3含量的变化而变化,如图 9 所示。由图可见,当CaCO3的质量分数为 10% (jCa-3)和 5%(jCa-2)时,改性后的吸附剂的吸附容量均得到一定程度的提高,吸附曲线与未改性的吸附剂相似,即它们的诱导期和快速吸附段的时间均相近。但是,当CaCO3的质量分数过高 (20%,jCa-4) 或者过低 (3%,jCa-1) 时,60 min 时吸附剂的吸附容量均比未添加CaCO3改性时的吸附剂的吸附容量低。这是因为,CaCO3含量过高(20%,jCa-4)时,吸附剂的活性组分含量减少,吸附剂的快速吸附段时间较短,导致吸附剂的表观吸附容量降低;而CaCO3含量过低(3%,jCa-1)时,改性吸附剂的诱导期较长,约10 min 后才进入快速吸附段,60 min 内未达到其饱和吸附容量。由此可知,吸附剂的吸附容量与CaCO3含量的关系不是单纯的递增或者递减的关系。
对图 9 的吸附容量~时间曲线进行微分,得到不同CaCO3含量的吸附剂的吸附速率曲线图,如图10 所示。由图可见,jCa-4 改性吸附剂吸附速率较慢,且快速吸附段时间较短,导致其吸附容量是最低的;jCa-1 改性吸附剂诱导期较长,前期吸附速率较低,10 min 后才进入快速吸附段,快速吸附段的时间跨度虽然较长,但吸附速率也较低,所以导致其吸附容量较低。结合图6 分析可知,主要是由于 jCa-1 改性吸附剂平均孔径和孔体积较小,CO2扩散速率较慢,导致其吸附速率较低。jCa-2 和jCa-3 改性吸附剂与jCa-0 未改性吸附剂的快速吸附段时间相近,但两者由于其孔体积较大,导致其在快速吸附段的吸附速率较快,所以其吸附容量也相对较高。
3.2.3 循环稳定性测试
不同 CaCO3含量的吸附剂的循环稳定性如图 11 所示,循环测试条件见2.4 节。由图可见,添加CaCO3后改性的吸附剂在20 次循环测试内的吸附容量衰减较小,说明CaCO3的添加有利于提高吸附剂的循环稳定性。但由于CaCO3是惰性组分添加剂,所以CaCO3的质量分数过高 (20%,jCa-4)时,吸附剂的稳定性虽然较好,但会减少吸附剂中活性组分的含量,导致其吸附容量降低。当 CaCO3的质量分数为 3 % ~10 % ( jCa-1、 jCa-2、jCa-3)时,吸附剂的初次吸附容量和20 次循环后的吸附容量均较高,说明 CaCO3的添加能提高吸附剂的循环稳定性。通过对比可以发现,CaCO3的含量有一最佳值,图11 表明,当CaCO3的质量分数为5%时制备的吸附剂吸附性能最好,初次吸附容量为0.63 g·g-1,20 次循环后吸附容量降为0.50 g·g-1。在相同的测试条件下,稳定性优于文献[16]中制备的吸附剂。
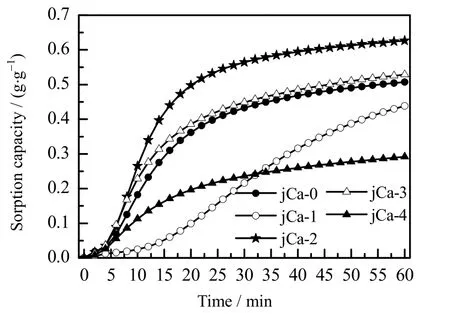
图9 不同CaCO3 含量的吸附剂的吸附容量曲线图Fig.9 Adsorption capacity curves of adsorbents with different CaCO3 contents

图10 不同CaCO3 含量的吸附剂的吸附速率曲线图Fig.10 Adsorption rate curves of adsorbents with different CaCO3 contents
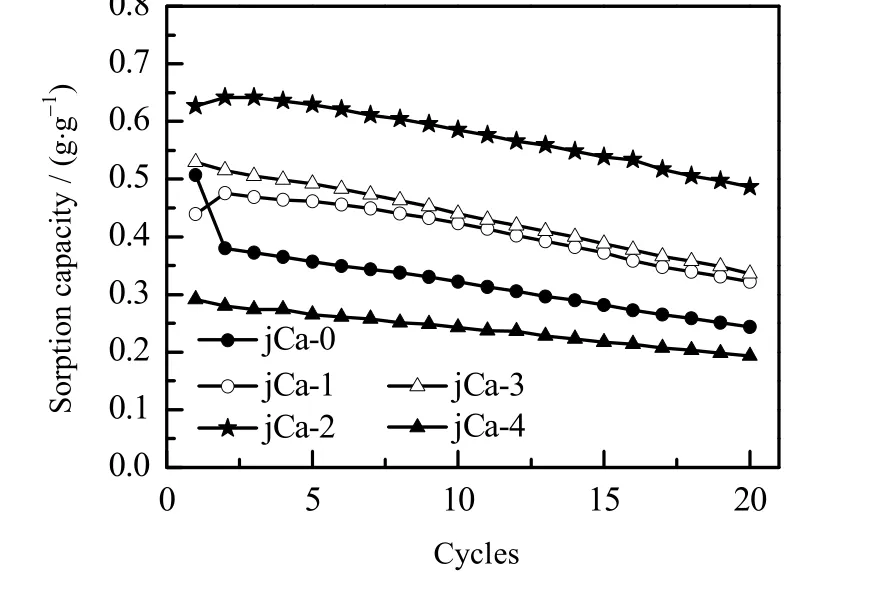
图11 不同CaCO3 含量的吸附剂的循环吸附容量Fig.11 Cyclic adsorption capacity of adsorbents with different CaCO3 contents
4 结 论
通过比较两种不同前驱体制备的MgO 基吸附剂发现,碱式碳酸镁制备的MgO 颗粒较小,吸附性能较好。采用塔曼温度高、呈碱性的CaCO3作为惰性添加剂,可以在一定程度上限制碱金属盐的高温下流动,进而提高吸附剂的循环稳定性。采用40% CO2气氛作为吸附气体、吸附温度为350 ℃、脱附温度为400 ℃,当CaCO3的质量分数为5% 时,吸附剂的初次吸附容量可达到0.63 g·g-1,20 次循环后吸附容量为0.50 g·g-1,体现了较好的循环稳定性。
符号说明:
B — 实测样品衍射峰半高宽度,rad
D — 晶粒垂直于晶面方向的平均厚度,nm
K — Scherrer 常数
γ — X 射线波长,0.154 056 nm
θ — 衍射角, rad
