SNP-STR遗传标记复合扩增体系的构建及法医学应用
2020-07-23
(四川大学华西基础医学与法医学院,四川 成都 610041)
SNP-STR遗传标记是由短串联重复序列位点与其相邻的单核苷酸多态性组成的单倍型。在STR位点的侧翼序列中存在SNP,可以利用等位基因特异性聚合酶链反应(allele-specific polymerase chain reaction,ASPCR)技术,又称扩增受阻突变系统(amplification refractory mutation system,ARMS)[1],实现SNP和STR等位基因的同步检测。其原理是针对STR重复结构侧翼序列中的SNP等位基因设计引物,将SNP所在的位置设置在引物3′端的3个碱基内,并通过在其两侧人为引入错配碱基使得引物与不同SNP等位基因的结合能力存在差异,从而实现对其中一种SNP等位基因的选择性扩增。ASPCR可在上千个细胞中检测出具有序列突变的细胞[2],在肿瘤细胞异质性[3]、母体外周血游离胎儿DNA分析[4]、亲子鉴定[5]以及混合DNA分析[6-8]等方面都有着广泛的应用价值。
混合DNA分析是法医遗传学领域的难题之一。在现行法医DNA分析体系中,以毛细管电泳为基础的STR图谱解析和似然率计算仍然是混合DNA分析的主要方法。在法医DNA样品中,两个体DNA混合是出现频率最高的混合类型,其中,量较大的个体DNA称为多数成分,量较少称为少数成分。多数成分与少数成分的比值称为混合比例(mixture ratio)。现行STR复合扩增体系在进行混合DNA分析时,其分析准确度主要受到混合DNA构成个体数以及混合比例的影响。一般而言,混合个体数越多、混合比例越大,则混合DNA的STR图谱解析难度越大。当混合比例超过10∶1时,少数成分的等位基因很难被检出或容易出现等位基因丢失的现象[9]。
基于ASPCR技术复合扩增SNP-STR[10]、SNPSNP[11]、插入缺失多态性(deletion/insertion polymorphism,DIP)-STR[12]遗传标记的方法可以提高混合DNA的分析效能。这是因为,当混合DNA样本中少数成分与多数成分的SNP或插入/缺失(insertion/deletion,InDel)等位基因不同时,可以通过SNP或InDel等位基因特异性扩增少数成分所特有的STR等位基因,从而避免多数成分对少数成分的竞争性扩增抑制,进而提高对混合DNA的分析效能。这种在少数成分与多数成分之间存在差异的SNP-STR或DIPSTR遗传标记又被称为信息遗传标记(informative marker)。之前的研究结果也验证了这一方法的有效性,WEI等[6]以及TAN等[7]分别构建了由8个和11个SNP-STR遗传标记构成的复合扩增体系来进行混合样本的分析,混合比例最高分别为20∶1和100∶1时,仍能得到信息遗传标记的完整分型。同时,WEI等[6]在观察单位点扩增时,混合比例最高可达1000∶1。
基于SNP-STR遗传标记在混合DNA分析中所具有的潜在优势,本研究将进一步探究SNP-STR遗传标记在法医DNA分析中的应用效能。SNP-STR遗传标记由于结合了SNP和STR的优势,能够利用少数成分所特有的SNP等位基因进行混合DNA的区分,以及使用STR等位基因在毛细管平台上进行分型[7]。因此,为增加其实际应用能力,本研究将在商业化试剂盒所使用的STR中筛选SNP-STR遗传标记,因为这些STR基因型信息与现有的规模化STR数据库兼容。其次,对于两个体DNA混合样本,SNP-STR遗传标记中出现的信息基因型概率[12]主要取决于SNP基因座。因此,本研究选择STR基因座重复结构两侧300个碱基范围内、最小等位基因频率大于0.1的SNP位点组成SNP-STR遗传标记,构建SNP-STR遗传标记复合扩增体系。在设计SNP等位基因特异性引物时,较此前研究使用“分别在引物3′端第二或第三位设计错配碱基”[6]或者“一强一稍弱”[7]的方法有所区别,本研究均在引物3′端的第三位碱基引入抑制引物-模板结合效果最强的错配碱基。为探究该体系的法医学效能,本研究利用四川汉族群体进行该体系的法医遗传学参数分析。此外,通过不同位点数量的复合扩增体系对不同混合比例的两个体DNA混合样本进行分析,探讨SNP-STR遗传标记复合扩增体系在混合DNA分析中的应用能力。
1 材料与方法
1.1 样本采集
根据知情同意原则,采集103名四川汉族无关健康个体的乙二胺四乙酸(ethylenediamine tetraacetic acid,EDTA)抗凝外周血样,置于-20℃保存备用。
1.2 DNA提取及定量
采用血液/细胞/组织基因组DNA提取试剂盒[天根生化科技(北京)有限公司]提取血液DNA,具体方法参照试剂盒说明书。采用Investigator Quantiplex试剂盒(德国Qiagen公司)对提取的DNA进行精确定量,具体方法参照试剂盒说明书。
1.3 SNP-STR遗传标记筛选
为增加本研究所构建体系与现有法医STR数据库的兼容性,本研究主要以商业化试剂盒中的STR基因座为待选位点,包括PowerPlex®21试剂盒(美国Promega公司)、Investigator IDplex Plus试剂盒(德国Qiagen公司)、EX22试剂盒(无锡中德美联生物技术有限公司)、AGCU 21+1荧光检测试剂盒(无锡中德美联生物技术有限公司)、MicroreaderTM23sp ID System(北京阅微基因技术有限公司)。
SNP位点的筛选使用基于Unix Shell语言的VCFtools脚本[13-14]在 1000 Genomes数据库[15]中完成,其筛选原则为:(1)SNP与STR重复结构的距离小于300bp;(2)SNP位点在中国汉族群体中的最小等位基因频率大于0.1;(3)SNP位点在中国汉族群体中为二等位基因。
1.4 等位基因特异性引物设计
利用Primer3进行在线引物设计(http://primer3.ut.ee/)。根据SNP位点设计相应的特异性扩增引物,引物3′端为SNP位点,每个SNP位点设计两条不同碱基末端的引物。为保证引物的扩增特异性,参照HUANG等[16]提出的错配碱基设计原则,在引物的3′端第三位碱基处设计错配碱基。引物长度为18~39 bp,GC含量为20%~60%,解链温度(melting temperature,Tm)为55~59℃,如果在扩增中观察到加A不全的现象,则在引物的5′端重新添加非人类基因组的特殊序列(GTTCTT)n。利用美国国立生物技术信息中心(National Center for Biotechnology Information,NCBI)上的Primer-BLAST工具检测引物特异性。具体引物序列如表1~2所示。
1.5 引物特异性验证
对于每一SNP-STR遗传标记均设计两对引物,其中3′端分别针对SNP位点的2个等位基因,5′端为相同的通用引物。每一SNP-STR遗传标记进行单位点扩增,并设置3个退火温度(51℃、54℃、57℃)来优化复合扩增体系。反应体系为:2×Premix TaqDNA聚合酶(日本TaKaRa公司)5 μL,10 μmol/L等位基因特异性引物 0.5 μL,10 μmol/L 通用引物0.5 μL,模板DNA 1 ng,无核酸酶水3 μL。PCR循环条件为:95℃预变性2 min;95℃变性30 s,51℃(或54℃和57℃)复性30 s,72℃延伸30 s,共35个循环;终延伸72℃ 5min。扩增片段采用8%聚丙烯酰胺凝胶电泳(polyacrylamide gel electrophoresis,PAGE)及银染进行分型检测。

表1 13个SNP-STR遗传标记的引物信息(Panel A)Tab.1 Primer information of the 13 SNP-STR markers(Panel A)

表2 13个SNP-STR遗传标记的引物信息(Panel B)Tab.2 Primer information of the 13 SNP-STR markers(Panel B)
利用Sanger测序验证电泳分型结果。利用Primer3对SNP-STR遗传标记设计Sanger测序引物,使其扩增产物将SNP和STR位点均包括在内,在长度上较SNP-STR等位基因特异性扩增产物更长。对于扩增片段过小(150bp左右)的引物对,在引物的5′端加上M13序列(F:TGTAAAACGACGGCCAGT;R:CAGGA AACAGCTATGACC)。
使用新设计的测序引物或M13引物进行测序,测序工作由英潍捷基(上海)贸易有限公司完成。测序引物序列见表3。

表3 测序引物序列信息Tab.3 Information of sequencing primers

续表3Continued tab.3
1.6 复合扩增体系及分析方法的建立
为尽可能减少引物间相互作用对复合扩增体系扩增效能的影响,本研究利用AutoDimer软件[17]对上述所有等位基因特异性引物和通用引物的引物间相互作用进行分析,根据分析结果对引物序列进行调整。经过筛选和调整,共得到13个SNP-STR遗传标记的26条等位基因特异性引物,并与13条各位点相应的通用引物共同构成26对等位基因特异性扩增引物,分成A和B两个复合扩增体系。
复合扩增反应体系为:2×Multiplex PCR Master Mix(德国 Qiagen 公司) 5 μL,Primer Mix 1 μL,模板DNA 1ng,无核酸酶水3μL。PCR扩增条件为:95℃预变性15 min;巢式扩增12个循环,包括95℃变性30s,57~51℃(每个循环降0.5℃)复性90s,72℃延伸30 s;20个循环扩增,包括95℃变性30 s,57℃复性90s,72℃延伸30s;终延伸72℃ 60min。
扩增产物在3130基因分析仪(美国Thermo Fisher Scientific公司)上进行毛细管电泳分离和基因型判读。总电泳体系为10μL,包括1μL扩增产物,8.9μL去离子甲酰胺,0.1μL内标AGCU Marker SIZ 500(无锡中德美联生物技术有限公司)。电泳介质为POP-7分离胶(美国Thermo Fisher Scientific公司),毛细管长36cm,进样电压3kV,进样时间8s,电泳电压9kV,时间30 min。采用GeneMapperTMIDv3.2软件(美国Thermo Fisher Scientific公司)进行基因分型。根据2800M标准品DNA以及已通过测序得到确定SNPSTR基因型分型的样本的电泳结果,获得各位点不同等位基因的电泳迁移参数,编写相应的panel文件和bin文件,建立等位基因分型标准品(ladder)。
1.7 群体遗传学参数分析
采用该体系对103例四川汉族个体样本进行基因分型。使用Arlequin 3.11软件对SNP-STR遗传标记进行Hardy-Weinberg平衡以及连锁不平衡(linkage disequilibrium,LD)检验。根据分型结果,统计SNPSTR单倍型数目以及不同SNP等位基因的单倍型数量和频率。使用PowerStats v12软件计算个体识别率(discrimination power,DP)、观察杂合度(observed heterozygosity,Ho)、典型父权指数(typical paternity index,TPI)以及非父排除率(probability of paternity exclusion,PE)。依据CASTELLA等[12]论文中的公式,计算信息基因型概率。同时,比较SNP-STR遗传标记复合扩增体系与相应STR基因座复合扩增体系的群体遗传学参数,包括累积个体识别率(cumulative discrimination power,CDP)、累积父权指数(combined paternity index,CPI)、累积非父排除率(cumulative probability of exclusion,CPE)以及平均观察杂合度。
1.8 SNP-STR遗传标记复合扩增体系的混合样本检测能力分析
1.8.1 单位点等位基因特异性扩增
为明确单个SNP-STR遗传标记对混合样本DNA的分析能力,本研究使用已知SNP-STR分型的DNA样品进行混合,混合比例为1 000∶1,其中少数成分DNA量为100pg。
1.8.2 复合扩增体系对混合样本的检测
为明确复合扩增体系对混合DNA的检测能力,以及体系中位点数目与检测能力的关系,本研究共采用4种不同位点数量的复合扩增体系,分别为13、10、8和6位点,其体系构成如表4所示。为确定混合样本中少数成分的模板量,首先利用4个不同的DNA样本来检测复合扩增体系的灵敏度,DNA模板量梯度设置为100、200、300、400、600和800pg。以样本的常规分型结果作为参照,以检出全部SNP-STR分型的最低模板量为检测限。
混合DNA由第103号样本(多数成分)和第76号样本(少数成分)构成,混合比例设置为100∶1和500∶1两个梯度。根据测序结果,已知此混合样本DNA中含有6个信息遗传标记,分别为s2325399C-D6S1043、rs1728369C-D16S539、rs9853910A-D3S3045、rs4847015T-D1S1656、rs17651965C-CSF1PO和rs28562816G-D6S477。其中,13位点体系中含6个信息遗传标记,10位点体系中含5个,8位点体系中含3个,6位点体系中含2个。
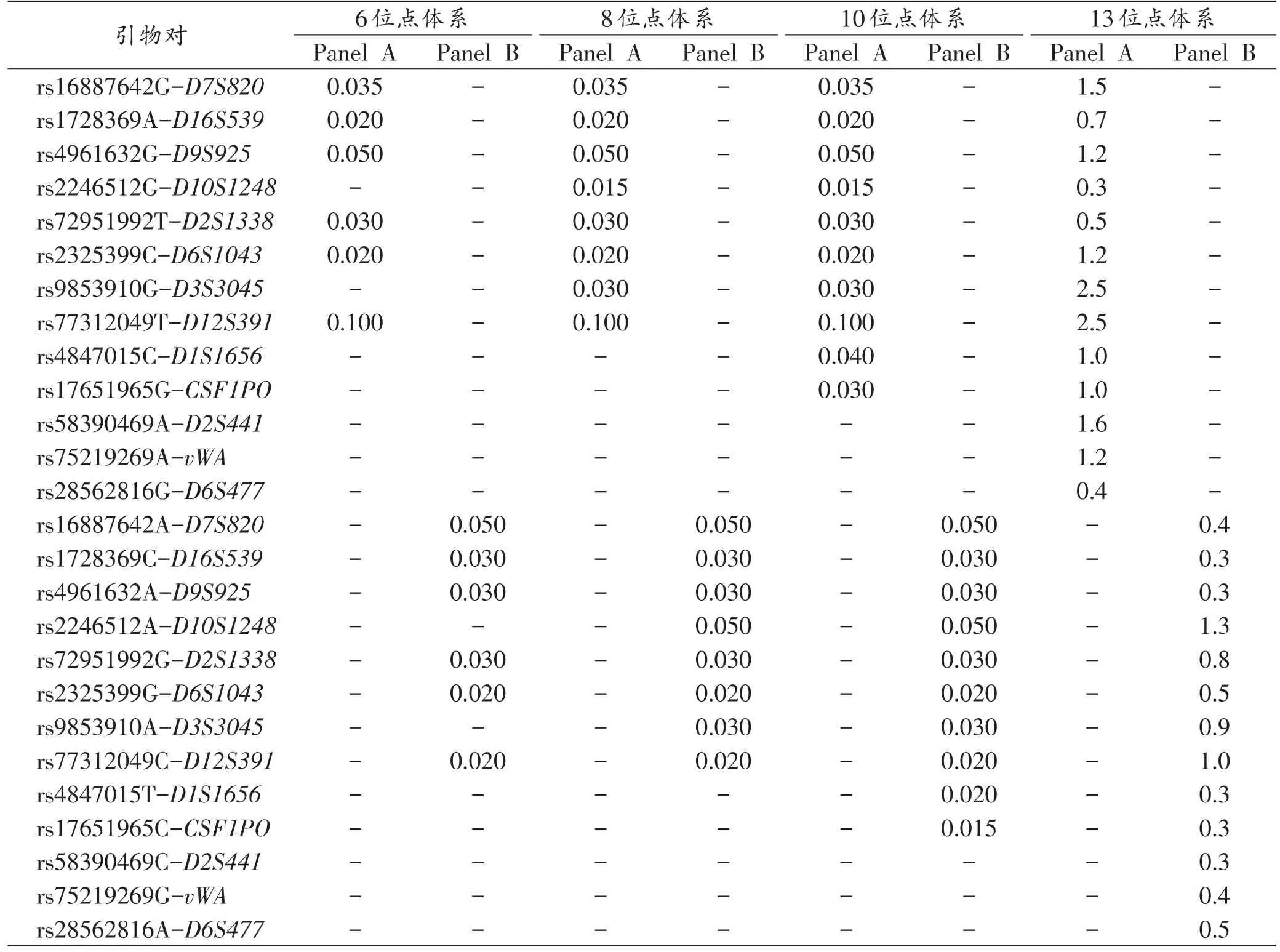
表4 6、8、10、13位点体系的组成和PCR引物终浓度Tab.4 The combinations and the primer concentrations of the 6,8,10 and 13 SNP-STR multiplex system(μmol)
2 结 果
2.1 引物特异性验证结果
本研究利用凝胶电泳及Sanger测序方法来验证等位基因引物的特异性。以rs4847015-D1S1656位点为例,图1为该位点等位基因特异性引物的电泳结果。从图中可以看出,只有5′端基于SNP等位基因C设计引物的泳道获得扩增产物,即该位点中的SNP基因型为C纯合子;STR有两条带,即该位点中的STR基因型为杂合子。不同退火温度的结果显示,57℃时的扩增特异性最好。此外,每一位点经过测序引物或M13引物扩增后采用Sanger测序进行验证,与电泳结果一致。
2.2 复合扩增体系的建立及灵敏度检测
本研究共筛选出13个SNP-STR遗传标记,并构建4种位点数目不同的复合扩增体系,分别为13、10、8和6位点。13位点体系的等位基因分型标准品建立结果如图2所示,2800M标准品DNA的分型结果如图3所示。
利用4个DNA样本来检测不同数目位点体系的灵敏度,每个样本的总模板量均设置为100、200、300、400、600和800pg。检测阈值设置为50RFU。结果表明,以检测到完整的SNP-STR分型为依据,13、10、8和6位点体系的复合扩增灵敏度分别为800、600、300和100pg。可以看出,体系的灵敏度随位点数目的增加而降低。
2.3 SNP-STR遗传标记复合扩增体系的群体遗传学参数
SNP-STR遗传标记之间均符合Hardy-Weinberg平衡。对位于同一染色体上的遗传标记进行配对连锁不平衡检验,未在此体系中检测到连锁不平衡。群体遗传学参数如表5所示。
在四川汉族人群中,各位点杂合度为0.76~0.88,信息基因型概率为 0.19~0.37,TPI为 2.06~4.29,PE为0.52~0.76。SNP-STR遗传标记复合扩增体系与相应STR基因座复合扩增体系的群体遗传学参数见表6,SNP-STR遗传标记复合扩增体系的CDP达0.999 999 999 999 999 968,CPI为 786 032.7,CPE 为0.9999985,平均Ho为0.82,均优于相应STR体系。
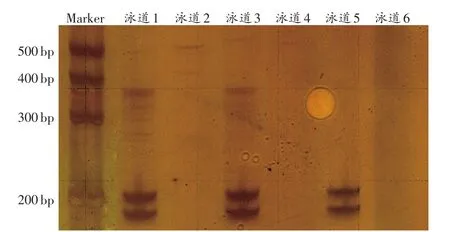
图1 rs4847015-D1S1656位点等位基因的特异性扩增PAGE结果Fig.1 The allele specific amplification products of rs4847015-D1S1656 based on polyacrylamide gel electrophoresis(PAGE)

图2 13位点体系的等位基因分型标准品Fig.2 The electrophoretogram of the standard allelic ladder of the 13 SNP-STR multiplex system

图3 2800M标准品DNA分型结果Fig.3 The typing result of the standard DNA 2800M

表5 SNP-STR复合扩增体系在四川汉族中的群体遗传学参数Tab.5 Forensic parameters of SNP-STR multiplex amplification system in Sichuan Han population(n=103)

表6 STR与SNP-STR复合扩增体系群体遗传学参数比较Tab.6 Comparison of the population genetic parameters between STR and SNP-STR multiplex amplification system
2.4 SNP-STR遗传标记复合扩增体系对混合样本的检测结果
利用单位点等位基因特异性扩增来判断每一位点对于混合DNA的检测能力,结果如图4所示,对于混合比例为1000∶1的混合DNA,每一SNP-STR遗传标记的等位基因特异性扩增体系均可对其中的信息遗传标记进行有效扩增并得到清晰的电泳峰型。
复合扩增体系的混合DNA检测能力由4种含不同位点数目的体系进行验证,结果如表7所示,当混合比例为100∶1时,随着位点数目的增加,实际检出的信息遗传标记均可保持在2/3以上(已知该混合DNA样本中共含6个信息遗传标记);当混合比例为500∶1时,实际检出的信息遗传标记数量较比例为100∶1时减少。

图4 等位基因特异性引物对混合DNA中少数成分的检测结果Fig.4 Genotyping results of the minor components in DNA mixture using allele-specific primers

表7 不同复合扩增体系分析混合样本的结果Tab.7 Detection of DNA mixture by different multiplex amplification systems
3 讨 论
本研究构建的SNP-STR遗传标记复合扩增体系基于商业化试剂盒中的STR基因座,其中9个来源于经过扩展的美国核心基因座(expanded U.S.core loci),7个来自欧洲标准位点(European Standard Set,ESS),所有基因座均已被广泛使用,可以实现该体系所获得的STR分型信息与现有法医DNA数据库兼容,便于其在法医学实践中推广使用。与13个STR基因座组成的体系相比,SNP-STR遗传标记复合扩增体系具有更高的DP和PE。在特殊亲子鉴定中,比如STR存在突变,或被检父与生父存在亲缘关系[5]时,为了明确父权需增加检测多态性好且稳定的遗传标记。由于SNP突变率低,而STR具有高多态性,若使用SNP-STR遗传标记,需要额外检测的遗传标记数目将明显下降,故SNP-STR遗传标记可以有效辅助亲缘关系的判断[5]。
在混合样本中,混合比例超过10∶1的混合DNA样本称为不平衡混合DNA。此类DNA在分析时容易出现等位基因丢失,主要源于PCR过程中多数成分与少数成分对同一引物的竞争性结合会抑制少数成分DNA的扩增。等位基因特异性扩增技术,在信息遗传标记存在的情况下,可以减少上述竞争性扩增抑制所导致的少数成分DNA分型丢失。但由于1个SNP-STR遗传标记具有2条序列上高度相似的特异性引物,且需要使用2种不同的荧光标记来区分SNP分型,共包含3条引物,因此相较于传统的STR复合扩增体系,SNP-STR遗传标记复合扩增体系单次检测遗传标记的数目有限,不同的引物组合方式对混合样本的检测能力具有较大的影响。WEI等[6]在单位点扩增时,将3条引物放入同一个PCR体系中扩增,混合比例最高为20∶1时可获得完整信息遗传标记分型;而将2条特异性引物分为2个体系扩增时,混合比例最高可达1000∶1。这可能是由于2条特异性引物序列高度相似,即使存在人为引入的错配碱基,也无法完全阻止错误引物与模板的结合。在此基础上,TAN等[7]通过在特异性引物3′端的第三位碱基引入不同的脱氧核苷酸来增大2条特异性引物的序列差异。然而,根据HUANG等[16]的实验结果,不同的错配碱基对于引物特异性的贡献存在差异。因此,本研究采取了在引物3′端的第3位使用抑制引物-模板结合效果最强的错配碱基,使2条特异性引物均实现最高水平的特异性。同时,为了避免同一位点序列高度相似的2条特异性引物相互竞争,本研究通过将复合扩增体系一分为二来增加体系对混合样本的检测能力。图4显示单位点扩增时,两个体混合样本混合比例达到1000∶1时依然可以得到少数样本的分型,这与WEI等[6]的研究结果一致。当进行多遗传标记复合扩增时,在混合比例为100∶1的情况下,随着位点数目的增加,实际检出的信息遗传标记数量均可保持在67%以上,且10位点与6位点体系能够实现信息遗传标记的全部检出,这验证了TAN等[7]的研究结果。此外,本研究进一步探索了复合扩增体系在混合比例达到500∶1时的混合分析能力,结果显示,部分信息遗传标记仍可被检出,且检测效能随着体系中位点数目的增加而降低,此现象可能与复合扩增体系中引物-引物和引物-模板间的相互作用有关。在今后的研究中,可考虑使用多个SNP-STR遗传标记复合扩增体系或者采用大规模并行测序(massively parallel sequencing,MPS)来进行SNP-STR分型。
综上所述,本研究通过等位基因特异性扩增技术,构建了13个SNP-STR遗传标记的复合扩增体系。该体系较单独使用相同STR基因座时具有更高的多态性,也具有更高的CDP和CPE。
