小分子ERK抑制剂的研究进展
2020-07-23梁停停王文杰郝思远何光超徐云根
梁停停,王文杰,郝思远,何光超,徐云根*
(1中国药科大学药物化学系,南京211198;2江苏省药物分子设计与成药性优化重点实验室,南京211198)
RAS-RAF-MEK-ERK 信号通路是一条广泛存在于哺乳动物细胞中的信号级联通路,在细胞的分化、存活、衰老和凋亡等细胞活动中发挥着极其重要的作用[1-2]。大量研究表明,在肿瘤的发展进程中,该信号通路处于持续异常激活状态。因此,以RAS-RAF-MEK-ERK 信号通路各成员蛋白为靶点的抗肿瘤药物的研发受到了人们的广泛重视[3]。目前,虽然尚无ERK 抑制剂成功上市,但是随着人们对ERK 结构与功能的深入研究,ERK 逐渐成为抗肿瘤药物研究的十分重要的靶点[4]。
1 MAPK 信号通路
丝裂原活化蛋白激酶(mitogen-activated protein kinases,MAPKs)信号通路可以将细胞外信号转导至细胞内,从而调节各种生物学功能[5]。在生长因子、氧化应激、炎症因子等细胞外信号的刺激下,该信号通路被激活,通过三级激酶级联的形式(MAPKKK→MAPKK→MAPK)传导细胞信号,调节哺乳动物细胞内相关基因的转录和表达,从而调控细胞的增殖、分化、凋亡、炎症反应[6]以及血管发育[7]等生物学功能。目前,在哺乳动物中,已经发现了5 种不同的MAPKs,分别为细胞外信号调节 激 酶(extracellular signal-regulated kinase,ERK)、C-Jun N 末端激酶(C-Jun N-terminal kinase,JNK)、p38 丝裂原活化蛋白激酶(p38 mitogen-activated protein kinase, p38MAPK)、ERK3/4 和ERK5[8](图1)。
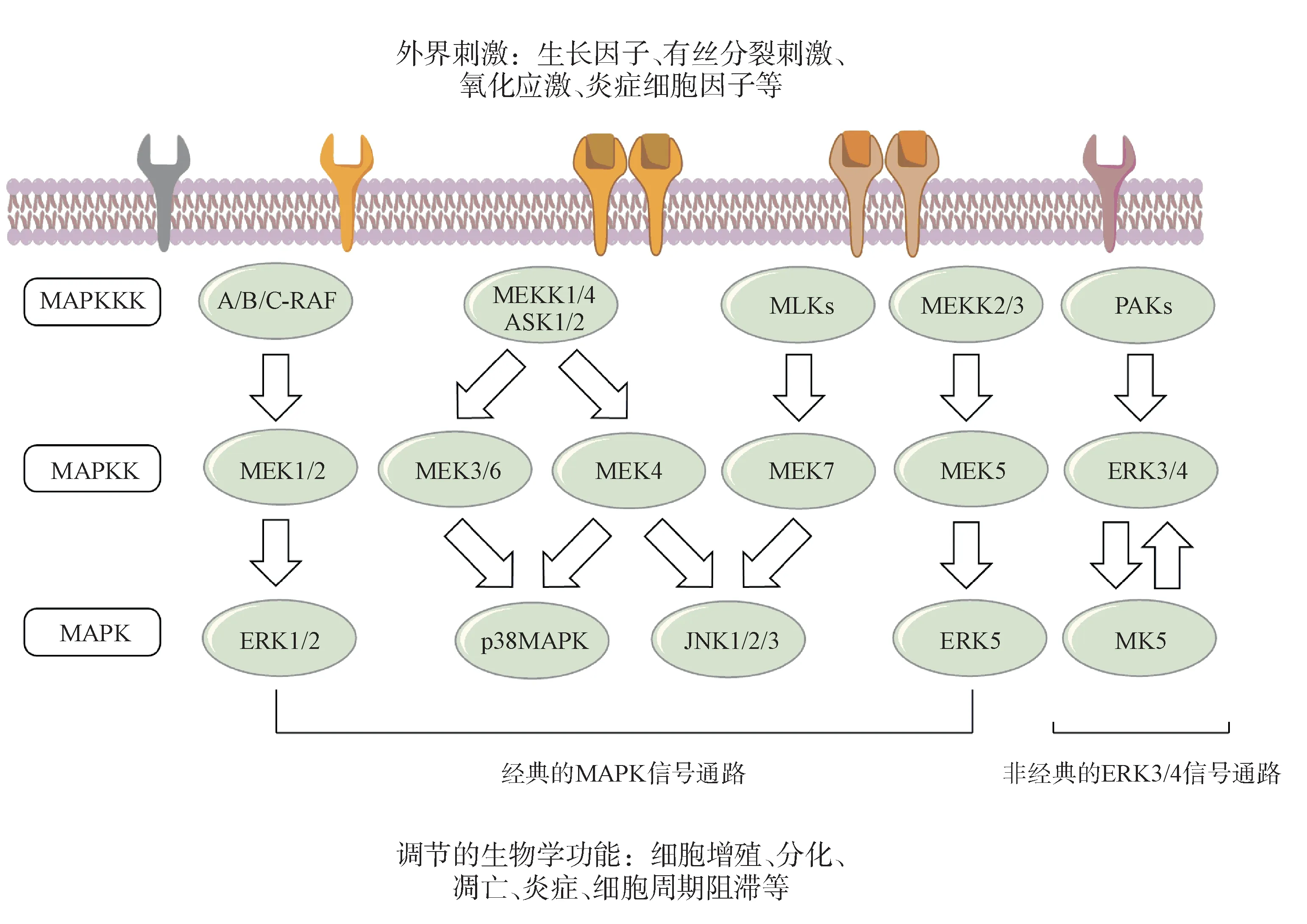
图1 哺乳动物体内的MAPKs信号传导通路
RAS-RAF-MEK-ERK 信号通路具有高度的保守性[9],其激活过程如下:首先,具有GTPase 活性的RAS 蛋白被上游的受体酪氨酸激酶(receptor tyrosine kinase,RTK)、细胞因子受体(cytokine receptor,CKR)和G 蛋白偶联受体(G protein coupled receptor,GPCR)等细胞表面受体激活[10],通过鸟苷酸交换因子(guanine nucleotide exchange factors,GEFs)使RAS 发生构象变化[11],从GDP 结合的非活化状态变为GTP 结合的活化状态[12]。然后,活化的RAS 通过与RAF 的N 端结构域结合,将RAF从细胞质招募到细胞膜上使其二聚化并激活[13]。RAF 是一个丝/苏氨酸蛋白激酶[14],活化的RAF 能够进一步与下游的MEK 蛋白结合,使其丝/苏氨酸残基磷酸化而导致MEK 的激活[10]。MEK1/2 是丝/苏氨酸和酪氨酸的双重特异性蛋白激酶,它能够磷酸化其下游的唯一底物erk1/2 的苏氨酸和酪氨酸残基使其活化。活化的ERK 进入细胞核,对其他激酶和转录因子等多种底物如RSK、CREB、Elk-1、NF-κB等进行磷酸化修饰[15],改变相关基因的表达,最终引起细胞周期进展以及细胞增殖、分化、代谢、凋亡等细胞行为的变化[16](图2)。
2 细胞外信号调节激酶(ERK)
2.1 ERK的结构与功能
与其他蛋白激酶类似,ERK 蛋白也具有由N端和C 端卷曲形成的双叶结构。其N 端由5 股反向平行的β 片层结构(β1~β5)、1 个αC 螺旋结构和一个甘氨酸富集环结构组成,C 端则由6 个保守的α 螺旋结构和4 股较短的β 片层结构(β6~β9)组成[17](图3)。

图2 RAS-RAF-MEK-ERK信号通路

图3 ERK2的二级结构(PDB:2OJJ)
几乎所有的哺乳动物体内都有ERK 蛋白的表达。到目前为止,ERK 蛋白被鉴定出有两个亚型:ERK1 和ERK2[18],二者在氨基酸序列上同源性高达84%[19]。以人源ERK 为例,ERK1 由379 个氨基酸残基组成,而ERK2 则由360 个氨基酸残基组成[20]。对于ERK1 和ERK2 的功能差异,目前主流的观点是ERK1 和ERK2 的生物学功能类似,只是两个亚型在整个ERK 蛋白活性中的贡献度不同[21]。但是Buscà 等[18]发现,在ERK1/2 基因破坏的小鼠中,ERK1/2表现出显著的表型差异:erk1-/-小鼠能够存活和正常繁殖,而ERK2被破坏则导致早期胚胎死亡。在对ERK1 和ERK2 之间的功能差异作进一步研究时,仅有少数实验揭示了ERK1和ERK2在功能上的微小差异,其具体的生物学机制仍有待探究。
目前,已知的ERK 底物数量多达上百种,主要分布在细胞膜、细胞质以及细胞核。ERK1/2 在细胞质底物包括RSK 家族蛋白激酶、磷蛋白磷酸酶、cAMP 磷酸二酯酶、胞浆磷脂酶A2、细胞骨架蛋白等[22-25],而ERK1/2 在细胞核内的底物大多是转录因子,主要是转录因子TCF 家族[26]。ERK 通过对不同底物的调节,从而发挥不同的生物学作用。
2.2 ERK信号通路在肿瘤发生发展中的作用
在约30%的肿瘤中,RAS-RAF-MEK-ERK 信号通路均处于异常激活状态。该信号通路中各成员蛋白的突变和异常表达在多种恶性肿瘤的发生发展过程中都发挥了重要作用。例如:在约1/3 的肿瘤中存在RAS的突变激活,其中KRAS的突变比率最高,占人类肿瘤总数的20%以上,在前列腺癌中的突变比率更是高达90%[27];在7%的肿瘤中含有BRAF 突 变[28],其 中 最 常 见 的 突 变 为BRAFV600E 突变,在黑色素瘤中的突变比率高达63%,在甲状腺乳头状癌中突变比率也有50%以上[29];在胃癌[30]、黑色素瘤[31]、结直肠癌[32]和肺癌[33]中均表现出MEK突变。
因此,针对RAS-RAF-MEK-ERK 信号通路研发的小分子靶向药物无疑在抗肿瘤药物研究领域占有突出地位[34]。
2.3 ERK抑制剂的优势
目前,已有多个BRAF 抑制剂和MEK 抑制剂被美国FDA 批准上市,用于治疗各种实体瘤,在临床治疗中取得了良好的效果。如:BRAF 抑制剂索拉非尼(sorafenib)、维罗非尼(vemurafenib)和达拉非 尼(dabrafenib);MEK 抑 制 剂 曲 美 替 尼(trametinib)、司美替尼(selumetinib)和考比替尼(cobimetinib)。虽然在患者接受治疗的初期,肿瘤的发展进程得到了有效控制,但是在长时间服用该类靶向药物后,肿瘤细胞不可避免的产生了获得性耐药。与RAS、BRAF 等在肿瘤细胞中的高突变率不同,到目前为止,ERK1/2的获得性突变在肿瘤细胞中几乎没有出现[35]。其原因可能是细胞中ERK1/2 能够调控激酶与转录因子等多种底物,一旦ERK1/2 发生耐药性突变,将会失去对这些底物的有效调控,影响正常细胞活动,无法保证细胞的存活。而且,越来越多的临床前研究结果显示,ERK 抑制剂比RAF 和MEK 抑制剂具有更好的效果。Qin 等[36]发现,选择性敲除ERK1/2 能够有效杀死A375 黑色素瘤细胞,同时增强A375 细胞对BRAF 抑制剂维罗替尼的敏感性;Hatzivassiliou等[37]发现,MEK 抑制剂耐药的细胞持续保持对MAPK 信号通路的依赖,其对选择性ERK1/2 抑制剂敏感,应用ERK抑制剂能够有效阻断细胞增殖。
因此,与抑制MAPK 信号通路中的上游靶点相比,抑制下游的ERK 同样能够起到阻断细胞信号转导的作用。更为重要的是,ERK 抑制剂能够克服肿瘤细胞对RAF 抑制剂和MEK 抑制剂的耐药性,在临床上具有更广泛的应用前景。
3 小分子ERK 抑制剂的研究进展
鉴于在肿瘤的发生发展过程中,MAPK 信号通路发挥了重要作用,而ERK 激酶又是MAPK 信号通路中十分关键的下游靶点。因此,选择性抑制ERK 能够阻断MAPK 信号通路,同时逆转上游靶点突变产生的耐药性。虽然目前尚无ERK1/2 抑制剂被正式批准上市,但是已经有一些小分子ERK 抑制剂处于临床或临床前研究阶段,下面将重点介绍小分子ERK抑制剂的研究进展。
3.1 处于临床研究阶段的ERK抑制剂
近年来,已有多个ERK 抑制剂进入临床研究,包括GDC-0994(1)、Ulixertinib(BVD-523)(2)、KO-947(3)、LY3214996(4)、MK-8353(5)、CC-90003(6)、LTT462等。
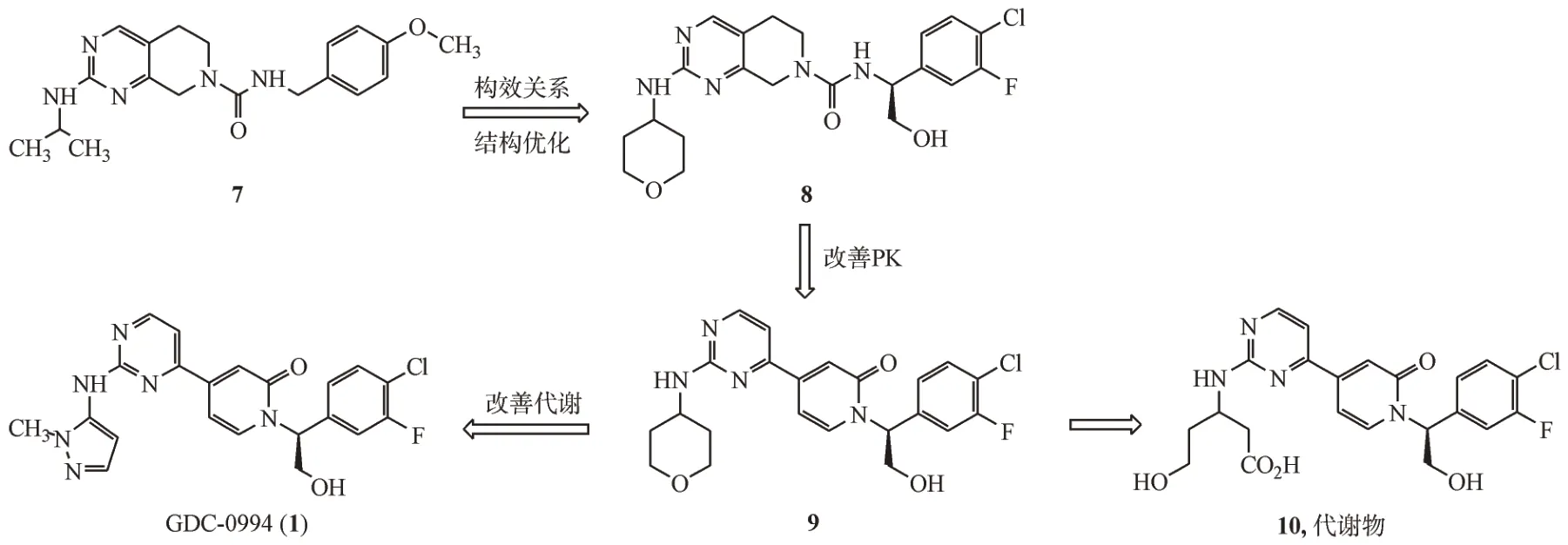
3.1.1 GDC-0994 GDC-0994(1)是由Genentech公司研发的口服的选择性ERK1/2 抑制剂,其对ERK1和ERK2的IC50分别为6.1和3.1 nmol/L。
2014 年,Genentech 公司通过高通量筛选得到四氢吡啶并嘧啶类化合物7,其对ERK2 的IC50为106 nmol/L。以化合物7 为先导化合物,经过结构优化得到化合物8,其对ERK2 的IC50提高到2 nmol/L,并且在人HCT116 结直肠癌的裸鼠移植模型中表现出较好的抗肿瘤效果[38]。但是,化合物8的药代动力学性质不佳,在动物体内清除率较高,口服生物利用度较低。为了改善化合物8的药代动力学性质,对其结构进行进一步优化,得到了化合物9[39]。但是,由于化合物9 中的四氢吡喃环在体内可以被代谢为羟基酸产物10,使得化合物9的预测给药量过大(>1 g/d)。为了进一步提高化合物9的活性和代谢稳定性,将结构优化集中在四氢吡喃环的改造上,最终得到化合物GDC-0994[40]。

分析GDC-0994与ERK2的共晶复合物(图4),可以发现2-氨基嘧啶结构能够与铰链区的Met108、Leu107 形成氢键作用,吡啶酮羰基能够通过水分子与门控残基Gln105 和催化残基Lys54 形成相互作用,羟甲基部分可以同时与Asp167 和Asn154 形成氢键作用,氟氯取代的苯基能够结合到甘氨酸富集环结构下方的疏水口袋。此外,5-氨基吡唑结构也能够与Lys114形成氢键相互作用。
动物实验结果表明,在HCT116 小鼠移植瘤模型中,GDC-0994 能够显著降低p90RSK 的磷酸化水平,抑制肿瘤组织的生长。在临床前的安全性评价试验中,与其他MEK 和ERK 抑制剂类似,GDC-0994 也表现出一些不良反应,包括大鼠体内由磷失调引发的软组织矿化、白蛋白水平下调和皮肤毒性、以及犬的胃肠道毒性等[40]。
目前,GDC-0994 用于治疗局部晚期或转移性实体瘤的Ⅰ期临床试验已经结束,进一步的临床研究暂无报道。

图4 化合物GDC-0994与ERK2的共晶复合物[40](PDB:5K4I)
3.1.2 Ulixertinib Ulixertinib (BVD-523,VRT752271,2)是由BioMed Valley Discoveries 公司研发的可逆型ATP 竞争性ERK1/2 抑制剂,其对ERK1 的Ki<0.3 nmol/L,对ERK2 的Ki= 0.04 nmol/L。无论是对活化的ERK2(pERK2)还是非活化的ERK2,Ulixertinib 均表现出强亲和力,但是对pERK2的亲和力要强于非活化的ERK2。
体外细胞实验结果显示,在BRAFV600E突变的UACC-62 黑色素瘤细胞中,Ulixertinib 浓度依赖性地抑制ERK1/2 的活性,使细胞周期被阻滞在G1期,从而发挥其抗增殖作用。此外,Ulixertinib还能诱导caspase-3/7 依赖性细胞凋亡通路的激活,促进肿瘤细胞的凋亡。动物实验结果表明,Ulixertinib 可以剂量依赖性抑制肿瘤的生长,同时对裸鼠体重基本没有影响[41]。更重要的是,在其他MAPK 信号通路抑制剂耐药的模型中,Ulixertinib仍然表现出抗增殖活性[42]。
目前,Ulixertinib 单独用药治疗晚期恶性肿瘤正处于Ⅱ期临床研究阶段(NCT01781429),用于治疗急性骨髓性白血病和骨髓增生异常综合征正处于Ⅰ/Ⅱ期临床研究阶段(NCT02296242)。在药物联用方面,Ulixertinib 与白蛋白结合型紫杉醇(nabpaclitaxel)和吉西他滨(gemcitabine)联合应用治疗转移性胰腺癌正处于Ⅰ期临床研究阶段(NCT02608229),与CDK4/6 抑制剂哌柏西利(palbociclib)联合应用治疗晚期胰腺癌和其他实体瘤正处于Ⅰ期临床研究阶段(NCT03454035)。
3.1.3 LY3214996 LY3214996(4)是由礼来公司研发的口服的ERK1/2 抑制剂(IC50=5 nmol/L),在BRAF和RAS突变的肿瘤细胞中可以抑制RSK1的磷酸化水平。2016 年作为治疗晚期实体瘤的药物进入Ⅰ期临床研究。其除了可以单独用药外,还可以与其他抗肿瘤药物联合使用,比如与CDK4/6抑制剂、PI3K/mTOR 抑制剂联用治疗RAS突变的非小细胞型肺癌[43]。
3.1.4 MK-8353 MK-8353(5)是 通 过 改 善SCH772984 的PK 而优化得到的(ERK1:IC50= 20 nmol/L;ERK2:IC50= 7 nmol/L)。SCH772984 是由Merck 公司通过基于亲和力的质谱高通量平台筛选、鉴定并通过结构优化得到的ATP 竞争性的ERK1/2 抑制剂。分析共晶结构,发现吲唑环与铰链区的Asp104 与Met106 形成至关重要的氢键作用,吡咯烷与Lys52 形成另一个关键的氢键,同时P-loop结构中的Tyr34残基翻转到ATP位点并堆叠到吡咯烷环的上方,导致P-loop 结构发生扭曲,Ploop 结合口袋被打开产生一个可以被甲基取代的三唑环占据的空腔。这些结论也表明,MK-8353对ERK 的高选择性主要是通过变构抑制的方式实现的[44]。因此,MK-8353 不仅能够ATP 竞争性地抑制ERK1/2 的催化活性,还能够诱导或稳定ERK 的构象,阻止其被MEK 激活[45]。到目前为止MK-8353 与帕姆单抗联用治疗晚期实体瘤已经进入Ⅰ期临床研究阶段[46]。2018 年,MK-8353 与司美替尼联用治疗晚期或转移性实体瘤也进入Ⅰ期临床研究[47]。
3.1.5 CC-90003 CC-90003(6)是不可逆的共价结合的选择性ERK1/2 抑制剂,主要用于治疗KRAS突变的肿瘤,2015年进入Ⅰ期临床研究。但是由于在Ⅰ期临床试验中,其最大耐受剂量(MTD)结果不令人满意,目前已经暂停了其临床试验[48]。经过分析共晶复合物,发现2-氨基嘧啶与铰链区形成至关重要的氢键作用,而丙烯酰胺基团则与Cys(ERK1:Cys183;ERK2:Cys164)形成不可逆的共价结合作用[49]。
3.2 尚处于临床前研究阶段的ERK抑制剂
除了上述介绍的已经进入临床的化合物,目前还有很多的ERK 抑制剂处于临床前阶段或生物活性评价阶段。这些化合物主要包括FR180204(11)、VTX-11e(12)、BL-EI-001(13)。

3.2.1 FR180204 Ohori 等[50]通过高通量筛选得到了ATP竞争性的ERK1/2抑制剂FR180204(11),对ERK1 和ERK2 的IC50分 别 为0.51 μmol/L 和0.33 μmol/L。
通过分析FR180204 与ERK2 的共晶复合物(图5),发现FR180204 占据了ERK2 的ATP 结合口袋。FR180204 结构中的吡唑并哒嗪环2′位的氮原子与Met108 形成氢键相互作用,3′位的氨基与Gln105 和Asp106 形成氢键作用,吡唑并吡啶环3 位的氮原子与Lys54 形成氢键作用。同时,FR180204结构中吡唑并哒嗪环与Leu156存在CHπ型相互作用,吡唑并吡啶环与Cys166 存在SH-π型相互作用。此外,在共晶复合物中还观察到,甘氨酸富集环采取了一种有利于与FR180204 结合的优势构象。这些独特的相互作用模式增强了FR180204 对ERK2 的选择性抑制,使其对p38α 的选择性高达30 倍,对MEK1、MKK4、IKKα 等其他激酶的选择性高100倍以上。

图5 FR180204与ERK2的共晶复合物(PDB:1TVO)
在Mv1Lu 貂肺上皮细胞中,FR180204 能够剂量依赖性地抑制TGFβ 诱导的AP-1 的激活,其IC50为3.1 μmol/L。动物实验结果表明,在CIA 小鼠模型中,FR180204 能够明显改善关节炎症状并恢复体重损失[51]。在登革病毒(DENV)感染的小鼠模型中,FR180204 通过抑制ERK1/2 的磷酸化,抑制肝细胞凋亡,减轻登革病毒诱导的肝损伤[52]。
目前,FR180204仍处于临床前的研究阶段,未见相关的临床试验报道。
3.2.2 VTX-11e Vertex 制药公司[53]通过高通量筛选得到具有吡唑并吡咯结构的化合物14,其对ERK 的Ki为2.3 μmol/L。通过对化合物14 与ERK共晶结构的分析(图6-A),发现化合物14结构中的吡唑环能够与ERK 铰链区的Met106、Asp104 形成至关重要的氢键作用。同时,酰胺键上的羰基能够与Lys52 形成氢键作用,吡咯环上的NH 能够与门控残基Gln103 形成氢键作用,苯基能够与甘氨酸富集环结构中的Val37 形成疏水作用。根据以上晶体结构信息,研究人员对化合物进行结构优化,发现具有(S)-苯基甘氨醇结构的化合物15 不仅增加了与Lys52 的疏水相互作用,羟甲基的引入还能与Asn152、Asp165形成氢键作用,从而有利于化合物与ERK 结合构象的稳定。此外,(S)-苯基甘氨醇结构上的卤素取代能够填充疏水空腔并与glycine-rich loop结构形成相互作用(图6-B)。这些结合上的优势,使得化合物15 表现出较好的ERK抑制活性(Ki=0.002 μmol/L)和选择性。但是,化合物15的细胞活性不够理想,在Colo205人结肠癌细胞增殖测试中,其IC50仅为0.54 μmol/L。
为了提高化合物的细胞活性,将化合物15 结构中与铰链区有相互作用的吡唑环替换为氨基嘧啶环,增强了化合物与铰链区的氢键作用,得到了先导化合物16。但是通过分析共晶结构(图6-C),发现与化合物15 相比,化合物16 吡咯环翻转了将近180°,导致了化合物对激酶的选择性大大降低。与化合物15相比,先导化合物16对GSK3、AuroraA和CDK2的抑制活性提高了250倍以上[54]。
为了提高先导化合物16 对ERK 的选择性,研究人员对苯胺部分和苯基甘氨醇部分进行结构优化,最终得到化合物VTX-11e(12)。VTX-11e 是有效的高选择性ERK2 抑制剂,其对ERK2 的Ki小于2.0 nmol/L,对GSK3、AuroraA 和CDK2 有200 倍以上的选择性,对受测的其他激酶的选择性更是大于500倍[54]。

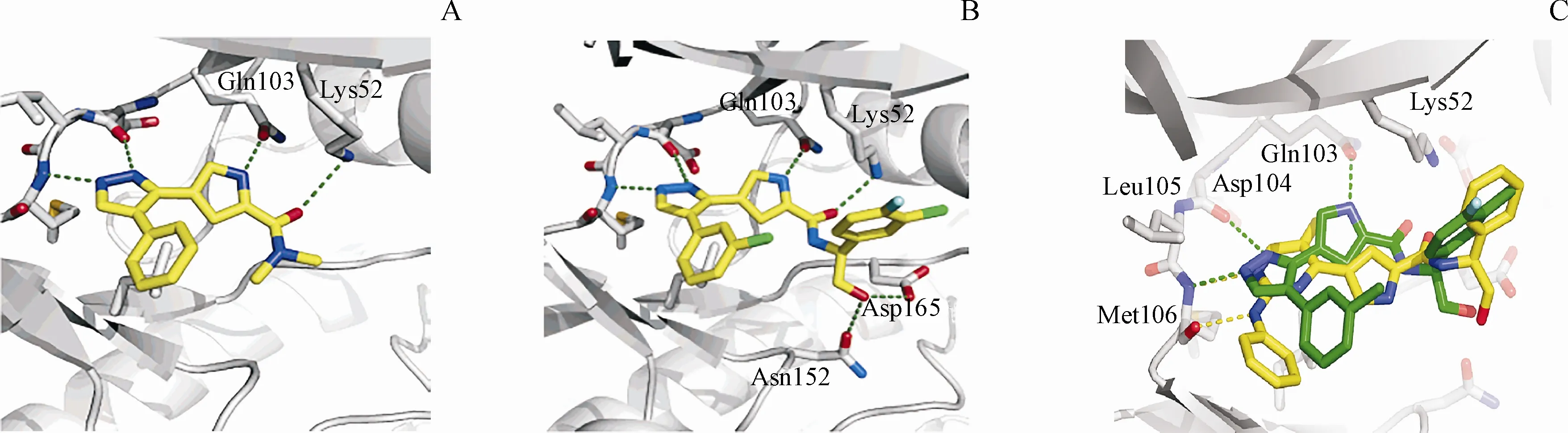
图6 化合物与ERK2的共晶复合物和叠合图
体外细胞研究结果显示,在HT29 人结肠癌细胞增殖测试中,VX-11e 的IC50为48 nmol/L。体内研究结果显示,在大鼠和小鼠中,VX-11e表现出较好的口服生物利用度。在对BRAF/MEK 抑制剂联合治疗和PI3K抑制剂单独治疗均有耐药性的人黑色素瘤移植的NSG 小鼠模型中,单独使用VX-11e能够抑制肿瘤组织生长;当与PI3K 抑制剂BKM120联用时,能够明显抑制肿瘤生长[55]。
目前,VX-11e 仍处于临床前的生物活性测试阶段。
3.2.3 BL-EI-001 BL-EI-001(13)是 由 清 华 大学、四川大学、沈阳药科大学联合开发的ERK 抑制剂,其设计策略是基于结构的药物设计,通过分子对接筛选DrugBank 和ZINC 得到了打分最高的11个化合物,然后通过生物活性测试确定先导化合物后再进行结构优化从而发现了BL-EI-001。通过对接发现疏水基团苯环与氨基酸残基Ile48、Val56、Ala69 和Met125 形成疏水作用,同时BL-EI-001可以与Lys71形成两个氢键作用,与Tyr53形成两个π-π作用。然而BL-EI-001 对肿瘤的抑制作用不是通过Ras/Raf/MEK1/2 通路,而是通过线粒体通路来发挥抗肿瘤作用[56]。
4 总结与展望
到目前为止,已有多个RAF 抑制剂和MEK 抑制剂应用于临床并取得了较好的肿瘤治疗效果。但是,在长期服用该类药物治疗肿瘤的过程中,不可避免的产生了耐药性问题。在对肿瘤耐药机制的研究中发现,原本被抑制的RAS-RAF-MEK-ERK信号通路被重新激活,其主要原因是RAF 和MEK的突变导致其对现有药物治疗的不敏感,从而引起患者肿瘤复发,治疗失败。本课题组通过骨架跃迁的药物设计策略,发现了一类异吲哚-1-酮类化合物对ERK 具有较好的抑制作用,在KRAS 和BRAF 突变的肿瘤中体现出良好的抗增殖活性[57]。因此,通过靶向抑制RAS-RAF-MEK-ERK 信号通路中关键的下游蛋白ERK,能够再次抑制重新活化的该信号通路,达到治疗耐药性肿瘤的目的。
虽然与RAF 和MEK 抑制剂深入的研究进展相比,ERK 抑制剂的研发较为滞后,但是以ERK 为靶点来特异性阻断RAS-RAF-MEK-ERK 信号通路的药物研发策略正越来越受到人们的重视。目前,已有多个ERK 抑制剂处于临床或临床前研究阶段。
随着ERK 抑制剂的不断研发以及临床研究的逐步深入,ERK 抑制剂有望成为继RAF 抑制剂和MEK 抑制剂之后的新一代MAPK 信号通路相关药物,克服RAF 抑制剂和MEK 抑制剂的耐药性问题,并在肿瘤的临床治疗方面产生积极而深远的影响。
