DNA羟甲基化调控动脉粥样硬化的研究进展
2020-07-21胡颖楚胡豪畅林少沂陈晓敏
胡颖楚,胡豪畅,林少沂,陈晓敏
综 述
DNA羟甲基化调控动脉粥样硬化的研究进展
胡颖楚1,2,胡豪畅1,林少沂1,陈晓敏1,2
1. 浙江大学宁波医院心血管内科,宁波 315000 2. 浙江大学医学院,杭州 310029
DNA羟甲基化作为一种表观遗传学修饰,对基因的表达调控起到了重要作用。近年来,越来越多的研究发现在心血管疾病中可见5-羟甲基胞嘧啶(5-hydroxymethylcytosine, 5hmC)和染色体10/11易位(ten-eleven translocation, TET)家族蛋白的异常改变,提示这些心血管疾病与DNA羟甲基化的调控密切相关。DNA羟甲基化水平与动脉粥样硬化常见的危险因素如衰老、性别、高血压和吸烟存在一定关联,并且和动脉粥样硬化发生过程中所涉及的免疫炎症反应以及内皮细胞和血管平滑肌细胞的功能相关。本文综述了DNA羟甲基化和TET家族蛋白对于动脉粥样硬化的作用机制及研究现状,以期为动脉粥样硬化的发生发展及诊断治疗提供表观遗传学方面的研究思路。
DNA羟甲基化;动脉粥样硬化;5hmC;TET;表观遗传学
心血管疾病(cardiovascular disease, CVD)在我国居民疾病死亡构成比中居首位,而动脉粥样硬化(atherosclerosis, As)作为CVD的主要致病原因,仅2016年我国因动脉粥样硬化性心血管疾病(atherosclerotic cardiovascular disease, ASCVD)而死亡的患者即有约240万人,占CVD死亡人数的61%[1]。目前治疗ASCVD的基本方法有药物治疗(如降血脂及抗血小板药物)和手术治疗(如经皮冠状动脉介入术、冠状动脉旁路搭桥术等)。虽然这些治疗手段能有效改善ASCVD的不良预后,但其死亡率仍呈逐年上升趋势,因此了解发生As的分子机制从而寻找新的治疗策略是目前急需解决的问题。
As的发病机制比较复杂,其早期病变主要涉及以下过程:当血管内皮细胞(vascular endothelial cells, VECs)的屏障功能被高血压、吸烟等As危险因素破坏后,单核细胞和淋巴细胞侵入血管内膜成为巨噬细胞,巨噬细胞吞噬氧化低密度脂蛋白(oxidized low-density lipoprotein, ox-LDL)后转变为泡沫细胞,同时生长因子和炎症因子大量释放并促进血管平滑肌细胞(vascular smooth muscle cells, VSMCs)的迁移和增殖,VSMCs吞噬脂质成为泡沫细胞的另一来源,泡沫细胞大量堆积从而形成As的脂质条纹。
As是由遗传因素与环境因素共同作用而引起的一种慢性炎症性疾病,而表观遗传学(epigenetics)作为一门探索遗传与环境因素相互作用的学科,近年来在As研究中逐渐受到重视。表观遗传学是指在不改变DNA序列的前提下对基因进行调控,导致基因表达水平发生可遗传的改变[2]。表观遗传学修饰包括DNA甲基化、组蛋白修饰和非编码RNA等,它们可参与调节基因组稳定性、X染色体失活等细胞的关键功能[3]。虽然同一个个体的细胞都携带大致相同的遗传信息,但不同种类细胞的基因表达谱却具有高度异质性,从而造成组织和器官的多样性,而这些基因表达的差异与表观遗传学机制有关[4]。表观遗传学具有一定的动态性和可逆性,这或许可为治疗As提供良好的药物靶点[5]。DNA羟甲基化(DNA hydroxymethylation)作为表观遗传学的一种类型,是指在染色体10/11易位(ten-eleven translocation, TET)家族蛋白催化下,DNA CpG岛的5-甲基胞嘧啶(5-methylcytosine, 5mC)氧化成为5-羟甲基胞嘧啶(5-hydroxymethylcytosine, 5hmC)[6]。DNA基因体或启动子区等部位发生羟甲基化修饰后能促进基因的表达,从而调节细胞发育和干细胞的多能性,而它的异常改变是许多疾病的发生基础[7]。越来越多的研究表明,衰老、性别、高血压和吸烟等As危险因素可通过影响某些靶基因的羟甲基化修饰水平,从而参与调控As的发生进展。本文从DNA羟甲基化与TET家族蛋白、DNA羟甲基化与As的危险因素和DNA羟甲基化与As的病变3个方面阐述了DNA羟甲基化在As发生发展过程中的作用和机制(图1),以期为As的预防、诊断和治疗提供一种新的表观遗传学方向。
1 DNA羟甲基化与TET家族蛋白
TET家族蛋白是一种5mC双加氧酶,包括TET1、TET2和TET3三种类型,它们均在DNA羟甲基化过程中起着关键作用。这3种TET蛋白的C末端都含一个富含半胱氨酸(cysteine-rich)和双链β螺旋(double-stranded beta-helix, DSBH)结构的CD结构域(cysteine-rich and DSBH regions, CD domain),它可与TET蛋白结构中的Fe2+和α-酮戊二酸结合,从而将5mC氧化为5hmC[8,9]。除CD结构域外,TET1和TET3的N末端还有一个CXXC锌指结构域,TET1的CXXC锌指结构域易与CpG岛结合,不仅能识别未修饰的胞嘧啶,还可识别5mC和5hmC[8]。此外,TET家族蛋白还可进一步氧化5hmC,并将其转化为5-甲酰胞嘧啶(5-formylcytosine, 5fC)和5-羧基胞嘧啶(5-carboxylcytosine, 5caC)[10]。
研究表明,TET家族蛋白中的3种蛋白在不同细胞中的表达不同,并参与了不同的生物学过程[11]。例如,TET1介导的羟甲基化可通过Wnt途径在肠道上皮的自我更新中发挥关键作用[12]。TET2在造血细胞中广泛表达,并可通过选择性调节、和等造血基因而参与红细胞的生成和分化[13]。TET3则高表达于受精卵中,并参与受精后父源基因组的重排[14]。此外,TET1和TET2通过特异性调控神经干细胞S期和G2/M期过程中基因的表达,在成年小鼠(Mus musculus)神经干细胞的增殖中发挥重要作用[15]。
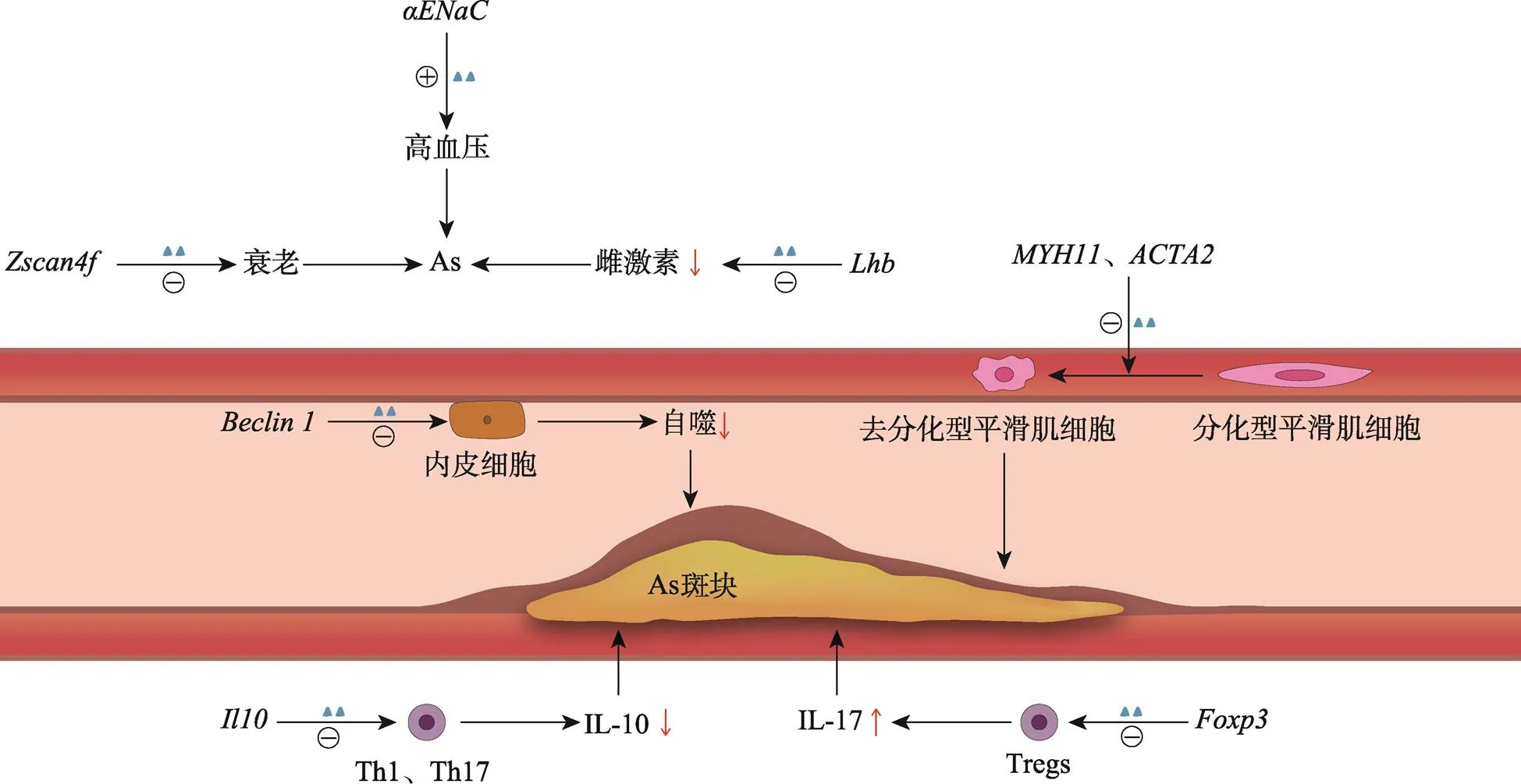
图1 DNA羟甲基化与部分靶基因在As斑块中的作用

除生物学功能外,TET家族蛋白还可通过改变DNA羟甲基化水平从而影响某些疾病的发生进展。目前许多研究表明DNA羟甲基化与肿瘤的发生发展存在密切关系。例如,TET1在结肠癌中显著下调,TET1低表达所致的低5hmC水平可促进肿瘤细胞的增殖[16];5hmC在黑色素瘤中的表达减少甚至缺失,而恢复正常的TET2水平可提高黑色素瘤模型的无瘤生存率[17];骨髓增生异常综合征(myelodysplastic syndromes, MDS)等血液系统恶性肿瘤中的错义突变可导致5hmC水平明显降低,并进一步造成了造血干细胞的异常分化[18]。还有研究表明,TET2和TET3可能在心脏早期发育过程中与心脏发育关键转录因子YY1 (Yin-Yang 1)结合,共同调控心脏发育,小鼠心脏特异性TET2和TET3缺失所致的低羟甲基化水平可导致胚胎致死性的心室非致密性心肌病[19]。由此可见,TET家族蛋白在心血管病中也发挥着重要作用。
2 DNA羟甲基化与动脉粥样硬化的危险因素
2.1 DNA羟甲基化与衰老
As多见于40岁以上的中老年人,衰老是As的一个独立危险因素。机体衰老涉及组织衰老和细胞衰老,其中细胞衰老的发生机制包括广泛的端粒缩短、DNA损伤增加、细胞增殖潜能的改变和细胞功能障碍等[20]。DNA羟甲基化近年来也被证明可能与As等多种早衰疾病有关。有研究表明,敲除上胚层样细胞(epiblast-like cells)的后,CpG位点的5hmC水平显著降低,同时可见基因簇的下调,而对维持端粒的稳定性至关重要[21]。进一步检测细胞周期中G1期端粒长度发现,端粒长度缩短的细胞比例明显增加[21]。由此可见,TET1及其介导的羟甲基化修饰可通过维持端粒的稳定性,从而延缓细胞的衰老进程。Johnson等[22]通过对年轻女性和老年女性外周血单核细胞的6650个不同羟甲基化区域(differentially hydroxymethylated regions, DhMRs)的检测发现,其中4664个DhMRs羟甲基化修饰水平与年龄呈正相关,最显著的区域出现在7号染色体上,该部位与丝氨酸/苏氨酸激酶17A (serine/threonine kinase 17A,)的内含子区重叠,STK17A属于死亡相关蛋白激酶(death-associated protein kinase, DAPK)家族成员,与细胞的凋亡有关[23];其余1986个DhMRs羟甲基化水平与年龄呈负相关,最显著的DhMRs为20号染色体的TOX内含子区,TOX2可修饰T淋巴细胞发育过程中的染色体结构,并与CD8+T细胞的衰竭相关[22]。此外,全基因组DNA羟甲基化水平随着机体的衰老表现出下降趋势[22]。
2.2 DNA羟甲基化与性激素
大量临床数据表明,绝经后的女性比绝经前的女性患ASCVD等心血管疾病的风险要大得多,这可能与雌激素的保护作用有关。研究发现,黄体生成素(luteinizing hormone beta polypeptide,)基因启动子区富含CpG岛,促性腺激素前体细胞中的下调可解除其对的抑制状态,进而在TET2的催化下,启动子区发生羟甲基化修饰,表达增加,黄体生成素分泌增多,从而促进卵巢的发育,提高雌激素的水平[24]。此外,虽然男性雄激素水平对As的作用仍有争议,但近年来许多研究表明雄激素水平下降反而会致As发病风险升高,这种雄激素对As的保护作用可能是由于睾酮在芳香化酶介导下转化为雌二醇,也可能是直接激活雄激素受体(androgen receptor, AR)所致[25]。基因敲除模型证明雄激素可能通过AR依赖途径抑制炎症因子和黏附分子的表达,抑制VSMCs的增殖和迁移,从而发挥其延缓As发生进展的作用[25]。研究发现,AR诱导的前列腺癌细胞miR-29a/b的表达可抑制的表达,低表达所致的低羟甲基化修饰水平可提高(forkhead box protein A1)增强子活性,进一步促进FOXA1相关雄激素调节因子与AR的结合,增加AR的敏感性并提高的转录活性[26]。以上研究表明,DNA羟甲基化可通过提高的表达水平从而促进雌激素的分泌;此外,雄激素调节通路的活性也受到了TET2及其介导的羟甲基化的负调控。
2.3 DNA羟甲基化与高血压
高血压患者As发病率较高,其原因可能与血流对血管壁的冲击导致血管重塑、内皮细胞受损、脂质沉积等有关[27]。盐敏感性高血压是原发性高血压的一种常见类型,其发病与高盐摄入等各种破坏水钠平衡因素所致的钠潴留和血浆容量增加有关,而α-上皮Na+通道(α-epithelial Na+channel, αENaC)可通过介导肾脏中Na+的重吸收从而参与其发病过程[28]。研究发现,醛固酮可使集合管主细胞的TET2发生聚集,从而使启动子区发生羟甲基化修饰,进一步促进的转录,导致集合管Na+的重吸收增加,机体水钠潴留,血压升高[29]。因此,DNA羟甲基化可通过αENaC从而参与盐敏感性高血压患者的发病过程。
2.4 DNA羟甲基化与吸烟
吸烟是As的一种常见危险因素,吸烟所产生的过氧化氢(H2O2)等活性氧可致VECs的氧化损伤及LDL的过氧化[30],从而通过氧化应激方式促进As的发生。H2O2可将鸟嘌呤氧化为8-氧代鸟嘌呤(8-oxoguanine, 8-oxoG),8-oxoG作为一种氧化损伤生物标记物,在吸烟者的肺组织中明显升高,且与日吸烟量有关[31,32]。在8-oxoG DNA糖基化酶1 (8-oxoguanine DNA glycosylase-1, OGG1)作用下,H2O2可进一步将TET1聚集到8-oxoG病变区,使邻近的启动子区CpG岛DNA发生羟甲基化修饰[32]。由此可见,吸烟可通过慢性氧化应激方式参与DNA的羟甲基化修饰,而慢性氧化应激作为As的发病机制之一,与As的发生进展密切相关。
3 DNA羟甲基化与动脉粥样硬化的病变
3.1 DNA羟甲基化参与调节免疫反应
As的病变过程涉及了免疫反应的参与,且与多种免疫细胞有关:ox-LDL激活内皮细胞后,血管内皮表面黏附分子如E-选择素和VCAM-1表达增加,它们与趋化因子CCL2等协同作用,吸引单核细胞、树突状细胞、T淋巴细胞等免疫细胞进入血管内膜,从而激活体内的固有免疫和特异性免疫系统[33]。固有免疫细胞如巨噬细胞吞噬ox-LDL后所形成的泡沫细胞是As斑块脂质条纹的来源,且巨噬细胞分泌的多种促炎因子和生长因子也能进一步促进As的病变。特异性免疫细胞如T淋巴细胞在As斑块中出现较早,其中Th1细胞可分泌促炎因子如IL-2和TNF-α,这些炎症因子能够激活巨噬细胞和VSMCs,增加斑块的不稳定性[34];调节性T细胞(regulatory T cells, Tregs)可分泌抑炎因子如IL-10和TGF-β,从而减轻As过程中的炎症反应,抑制斑块的形成[35]。
IL-10作为一种抑炎因子,可通过抑制巨噬细胞的活化以及抑制促炎因子的分泌和基质金属蛋白酶的合成,起到减少泡沫细胞形成、稳定As斑块的作用[36]。敲除Th17细胞的后,可检测到10位点CNS2区的5hmC水平下调,从而导致10表达下降,IL-10分泌减少[37]。与Tet2T淋巴细胞相比,Tet2T淋巴细胞表现出明显的IL-10抑制作用。此外,敲除Th1细胞的也可发现类似结果[37]。由此可见,TET2及其介导的羟甲基化修饰可促进辅助性T细胞表达抑炎因子,从而发挥抗As的作用。
Foxp3 (forkhead box-p3)是Tregs最重要和特异的转录因子,参与了Tregs的生长发育过程[38]。有研究表明,T细胞特异性的双基因敲除,可导致Tregs的上游增强子区域DNA羟甲基化水平受损,表达减少,从而增加Tregs的不稳定性并降低其抑制活性,导致Tregs转化为分泌IL-17的CD4+T细胞,使IL-17分泌增加[39]。而As作为一种慢性炎症性疾病,其病理变化中VECs的破坏、免疫细胞的聚集、VSMCs的迁移、斑块的破裂过程均有IL-17等促炎因子的参与。因此,DNA羟甲基化修饰可通过调节Tregs的分化进而参与As的病变。
以上研究表明,As过程中的免疫反应受到DNA羟甲基化修饰的调节,羟甲基化修饰水平的下调可影响免疫细胞的分化和功能,并进一步激活炎症反应,提高促炎因子、抑制抑炎因子,共同促进As的发生进展。
3.2 DNA羟甲基化参与调节血管内皮细胞的程序性细胞死亡
当VECs的屏障功能受到破坏后,单核细胞易于通过受损的内皮进入血管内膜,同时暴露的内皮下组织激活血小板,活化的血小板释放许多细胞因子促进SMCs的增殖,从而加速As的发展。研究表明,DNA羟甲基化可通过调节VECs的程序性细胞死亡(programmed cell death, PCD)如自噬(autophagy)和焦亡(pyroptosis)过程,从而参与As的发生进展。
自噬是普遍存在于真核细胞中的一种维持细胞健康的自我调节方式,它通过溶酶体来降解异常的蛋白质和损伤的细胞器,自噬过程受损可致脂质代谢障碍和VECs受损[40]。Beclin 1作为形成自噬小体的关键调节因子,它可通过溶酶体自噬途径来介导泛素化蛋白聚集体和线粒体的降解[41]。有研究发现,过表达ox-LDL处理后的人脐静脉内皮细胞后,基因启动子区发生羟甲基化修饰,Beclin 1释放增加,且VECs自噬作用增强;而在过表达的ApoE小鼠As斑块中,同样可检测到5hmC水平的升高以及Beclin 1的增加,同时As斑块面积明显减少,这是通过调节Beclin 1依赖的自噬过程而实现的[41]。此外,血流动力学的变化如在血流低切应力条件下,也可发现血管内皮细胞羟甲基化修饰水平下调所致的表达下降[42]。
细胞焦亡又称细胞炎性坏死,它以质膜破裂、胞浆内容物和炎性介质释放到胞外从而激活强烈的炎症反应为特征[43]。焦亡是一种依赖于炎性细胞内蛋白酶caspase-1的PCD途径,高血脂等As危险因素被VECs的caspase-1炎性小体途径感知后,即可使VECs发生焦亡[43]。研究发现,ox-LDL上调VECs miR-125a-5p的表达水平后,后者通过作用于的3ʹ非编码区(3ʹuntranslated region, 3ʹ-UTR),抑制了TET2蛋白的表达,从而下调了VECs 5hmC水平,而焦亡相关蛋白caspase-1活性升高[44]。同时,VECs焦亡过程中所释放的炎症因子及黏附分子如ICAM-1和VCAM-1,促进了单核细胞向内膜的迁移,从而推动了As的发生发展。然而,VECs焦亡过程中DNA羟甲基化修饰的具体调控位点及机制仍有待于进一步研究。
3.3 DNA羟甲基化参与调节血管平滑肌细胞的表型转化
在As病变过程中,VSMCs可发生表型转化,即由分化型(又称收缩型)转变为去分化型(又称合成型)。分化型VSMCs可维持血管的收缩性,而去分化型可增加VSMCs的增殖性和迁移性,导致血管顺应性降低和管腔狭窄,从而促进As斑块的形成[45]。研究发现,敲除VSMCs的后,VSMCs的分化标志物肌球蛋白重链11 (smooth muscle-myosin heavy chain,)及平滑肌肌动蛋白α2 (smooth muscle actin,)启动子区5hmC表达下降,MYH11和ACTA2分泌减少,VSMCs发生向去分化型的转化,并进一步致血管内膜增生面积增加[46]。进一步的体外试验也表明,用雷帕霉素诱导VSMCs分化后,可见分化基因的5hmC表达增加;反之用血小板衍生生长因子BB (platelet derived growth factor BB, PDGF-BB)诱导VSMCs去分化后,分化基因的TET2及5hmC表达下降[46]。由此可见,DNA羟甲基化修饰参与了As过程中VSMCs表型转化的调控过程,VSMCs分化基因羟甲基化修饰水平的上调可致其表达增多,从而抑制了VSMCs增殖和迁移,并强烈拮抗内膜增生,有效控制这种表型的转化可能对As起到预防作用。
4 结语与展望
近年来,DNA羟甲基化与心血管病的研究取得了一定进展,多项研究发现DNA羟甲基化与衰老、性别、高血压和吸烟等As的危险因素有关,并在As病变过程中调节了免疫细胞的分化和炎症因子的分泌,介导了VECs的程序性细胞死亡如自噬和焦亡,还参与调节分化型VSMCs和去分化型VSMCs之间的表型转化,这为As的发生发展提供了一种新的表观遗传学思路。
但是,目前大部分研究只是建立于DNA羟甲基化所调控的基因基础上,而没有深入到具体位点,且以往的测序方法无法区分5mC和5hmC,这为进一步研究DNA羟甲基化造成了一定困难[47]。一些新的5hmC测序技术的出现,如TET依赖的重亚硫酸盐测序(Tet-assisted bisulfite sequencing, TAB-seq)技术和5hmC特异性的寡核苷酸测序(5hmC-specific tethered oligonucleotide-primed sequencing, hmTOP- seq)技术[48,49],为人们进一步寻找具体位点提供了技术支持,也为心血管疾病的诊断提供了新的思路。选择性化学标记(selective chemical capture, hmC- Seal)技术证明,源自循环游离DNA (circulating cell- free DNA, cfDNA)的5hmC标记物可对冠心病进行诊断和预测,且表现出与肌钙蛋白I相当的诊断性能[50]。此外,最近有研究发现,随着颈动脉斑块严重程度的增加,外周血单个核细胞(peripheral blood mononuclear cells, PBMCs)的5hmC水平呈逐渐上升趋势,5hmC联合颈动脉斑块Crouse评分或许可作为诊断冠状动脉粥样硬化的生物标志物[51]。
迄今为止,表观遗传学药物如DNA甲基转移酶(DNA methyltransferases, DNMT)抑制剂阿扎胞苷和地西他滨已被广泛用于MDS等血液系统恶性肿瘤的治疗,它们可通过DNA去甲基化机制重新编程抑癌基因,从而阻止肿瘤细胞分裂,发挥抗肿瘤的作用[52,53]。在心血管方面,有研究表明,血管扩张剂肼曲嗪可通过抑制DNMT进而降低心肌细胞Ca2+-ATP酶2a (sarcoplasmic reticulum Ca2+-ATPase2a, SERCA2a)的甲基化水平,从而调节Ca2+的分泌,发挥其治疗心衰的作用[54]。还有研究证明,抑制肾上腺素β1受体(adrenergic beta1 receptor,)启动子的甲基化水平可以提高美托洛尔的降压疗效[55]。然而,目前对于DNA羟甲基化的研究还处在初步阶段,用于治疗心血管病的羟甲基化药物极少被报道,因此探索更有效的药物靶点从而研发特异性的羟甲基化药物可成为我们下一阶段的研究目标。
尽管DNA羟甲基化在As中的作用已取得了一定的成果,但DNA羟甲基化修饰调控As的具体分子机制及可能的治疗靶点和后续的临床应用仍需未来不断地探索。As作为一种遗传和环境因素共同作用的慢性疾病,其发生机制复杂且影响因素多样,相信随着人类表观基因组工程的发展,DNA羟甲基化修饰有望为As的预防、诊断和治疗提供新的靶点和思路。
[1] Zhao D, Liu J, Wang M, Zhang XG, Zhou MG. Epidemiology of cardiovascular disease in China: current features and implications., 2019, 16(4): 203–212.
[2] Zhang JW, Xu Q, Li GL. Epigenetics in the genesis and development of cancers., 2019, 41(7): 567–581.张競文, 续倩, 李国亮. 癌症发生发展中的表观遗传学研究. 遗传, 2019, 41(7): 567–581.
[3] Ho SM, Tang WY. Techniques used in studies of epigenome dysregulation due to aberrant DNA methylation: an emphasis on fetal-based adult diseases., 2007, 23(3): 267–282.
[4] van der Harst P, de Windt LJ, Chambers JC. Translational perspective on epigenetics in cardiovascular disease., 2017, 70(5): 590–606.
[5] Wierda RJ, Geutskens SB, Jukema JW, Quax PH, van den Elsen PJ. Epigenetics in atherosclerosis and inflammation., 2010, 14(6A): 1225–1240.
[6] Zhang YX, Gao KR, Yu SY. Progress of research on 5-hydroxymethylcytosine., 2012, 34(5): 509–518.张燕霞, 高可润, 禹顺英. 5-羟甲基胞嘧啶的研究进展. 遗传, 2012, 34(5): 509–518.
[7] Richa R, Sinha RP. Hydroxymethylation of DNA: an epigenetic marker., 2014, 13: 592–610.
[8] Tan L, Shi YG. Tet family proteins and 5-hydroxymethylcytosine in development and disease., 2012, 139(11): 1895–902.
[9] Wang J, Zhang KX, Lu GZ, Zhao XH. Research progress on 5hmC and TET dioxygenases in neurodevelopment and neurological diseases., 2017, 39(12): 1138–1149.王建, 张凯翔, 芦国珍, 赵湘辉. 5-羟甲基胞嘧啶及其TET氧合酶在神经系统发育和相关疾病中的研究进展. 遗传, 2017, 39(12): 1138–1149.
[10] Ito S, Shen L, Dai Q, Wu SC, Collins LB, Swenberg JA, He C, Zhang Y. Tet proteins can convert 5-methylcytosine to 5-formylcytosine and 5-carboxylcytosine., 2011, 333(6047): 1300–1333.
[11] Ito S, D'Alessio AC, Taranova OV, Hong K, Sowers LC, Zhang Y. Role of Tet proteins in 5mC to 5hmC conversion, ES-cell self-renewal and inner cell mass specification., 2010, 466(7310): 1129–1133.
[12] Kim R, Sheaffer KL, Choi I, Won KJ, Kaestner KH. Epigenetic regulation of intestinal stem cells by Tet1- mediated DNA hydroxymethylation., 2016, 30(21): 2433–2442.
[13] Ge L, Zhang RP, Wan F, Guo DY, Wang P, Xiang LX, Shao JZ. TET2 plays an essential role in erythropoiesis by regulating lineage-specific genesDNA oxidative demethylation in a zebrafish model., 2014, 34(6):989–1002.
[14] Gu TP, Guo F, Yang H, Wu HP, Xu GF, Liu W, Xie ZG, Shi LY, He XY, Jin SG, Iqbal K, Shi YG, Deng ZX, Szabo PE, Pfeifer GP, Li JS, Xu GL. The role of Tet3 DNA dioxygenase in epigenetic reprogramming by oocytes., 2011, 477(7366): 606–610.
[15] Shimozaki K. Ten-eleven translocation 1 and 2 confer overlapping transcriptional programs for the proliferation of cultured adult neural stem cells., 2017, 37(6): 995–1008.
[16] Neri F, Dettori D, Incarnato D, Krepelova A, Rapelli S, Maldotti M, Parlato C, Paliogiannis P, Oliviero S. TET1 is a tumour suppressor that inhibits colon cancer growth by derepressing inhibitors of the WNT pathway., 2015, 34(32): 4168–4176.
[17] Lian CG, Xu YF, Ceol C, Wu FZ, Larson A, Dresser K, Xu WJ, Tan L, Hu YQ, Zhan Q, Lee CW, Hu D, Lian BQ, Kleffel S, Yang YJ, Neiswender J, Khorasani AJ, Fang R, Lezcano C, Duncan LM, Scolyer RA, Thompson JF, Kakavand H, Houvras Y, Zon LI, Mihm MC Jr, Kaiser UB, Schatton T, Woda BA, Murphy GF, Shi YG. Loss of 5-hydroxymethylcytosine is an epigenetic hallmark of melanoma., 2012, 150(6): 1135–1146.
[18] Ko M, An J, Pastor WA, Koralov SB, Rajewsky K, Rao A. TET proteins and 5-methylcytosine oxidation in hematological cancers., 2015, 263(1): 6–21.
[19] Fang SH, Li J, Xiao Y, Lee M, Guo L, Han W, Li TT, Hill MC, Hong TT, Mo W, Xu R, Zhang P, Wang F, Chang J, Zhou YB, Sun DQ, Martin JF, Huang Y. Tet inactivation disrupts YY1 binding and long-range chromatin interactions during embryonic heart development., 2019, 10(1): 4297.
[20] Wang JC, Bennett M. Aging and atherosclerosis: mechanisms, functional consequences, and potential therapeutics for cellular senescence., 2012, 111(2): 245–259.
[21] Khoueiry R, Sohni A, Thienpont B, Luo X, Velde JV, Bartoccetti M, Boeckx B, Zwijsen A, Rao A, Lambrechts D, Koh KP. Lineage-specific functions of TET1 in the postimplantation mouse embryo., 2017, 49(7): 1061–1072.
[22] Johnson ND, Huang LX, Li RH, Li Y, Yang YC, Kim HR, Grant C, Wu H, Whitsel EA, Kiel DP, Baccarelli AA, Jin P, Murabito JM, Conneely KN. Age-related DNA hydroxymethylation is enriched for gene expression and immune system processes in human peripheral blood., 2020, 15(3): 294–306.
[23] Ozeki M, Salah A, Aini W, Tamaki K, Haga H, Miyagawa- Hayashino A. Abnormal localization of STK17A in bile canaliculi in liver allografts: an early sign of chronic rejection., 2015, 10(8): e0136381.
[24] Yosefzon Y, David C, Tsukerman A, Pnueli L, Qiao S, Boehm U, Melamed P. An epigenetic switch repressing Tet1 in gonadotropes activates the reproductive axis., 2017, 114(38): 10131–10136.
[25] Takov K, Wu JX, Denvir MA, Smith LB, Hadoke PWF. The role of androgen receptors in atherosclerosis., 2018, 465:82–91.
[26] Takayama K, Misawa A, Suzuki T, Takagi K, Hayashizaki Y, Fujimura T, Homma Y, Takahashi S, Urano T, Inoue S. TET2 repression by androgen hormone regulates global hydroxymethylation status and prostate cancer progression., 2015, 6: 8219.
[27] Hurtubise J, McLellan K, Durr K, Onasanya Q, Nwabuko D, Ndisang JF. The different facets of dyslipidemia and hypertension in atherosclerosis., 2016, 18(12): 82.
[28] Pavlov TS, Staruschenko A. Involvement of ENaC in the development of salt-sensitive hypertension., 2017, 313(2): F135–F140.
[29] Yu ZY, Kong Q, Kone BC. Aldosterone reprograms promoter methylation to regulate αENaC transcription in the collecting cuct., 2013, 305(7): F1006– F1013.
[30] Siasos G, Tsigkou V, Kokkou E, Oikonomou E, Vavuranakis M, Vlachopoulos C, Verveniotis A, Limperi M, Genimata V, Papavassiliou AG, Stefanadis C, Tousoulis D. Smoking and atherosclerosis: mechanisms of disease and new therapeutic approaches., 2014, 21(34): 3936–3948.
[31] Ringh MV, Hagemann-Jensen M, Needhamsen M, Kular L, Breeze CE, Sjoholm LK, Slavec L, Kullberg S, Wahlstrom J, Grunewald J, Brynedal B, Liu Y, Almgren M, Jagodic M, Ockinger J, Ekstrom TJ. Tobacco smoking induces changes in true DNA methylation, hydroxymethylation and gene expression in bronchoalveolar lavage cells., 2019, 46: 290–304.
[32] Zhou XL, Zhuang ZH, Wang WT, He LF, Wu H, Cao Y, Pan FY, Zhao J, Hu ZG, Sekhar C, Guo ZG. OGG1 is essential in oxidative stress induced DNA demethylation., 2016, 28(9): 1163–1171.
[33] Hansson GK, Hermansson A. The immune system in atherosclerosis., 2011, 12(3): 204–212.
[34] Wu MY, Li CJ, Hou MF, Chu PY. New insights into the role of inflammation in the pathogenesis of atherosclerosis., 2017, 18(10): 2034.
[35] Hansson GK. Inflammation, atherosclerosis, and coronary artery disease., 2005, 352(16): 1685–1695.
[36] Han XB, Boisvert WA. Interleukin-10 protects against atherosclerosis by modulating multiple atherogenic macrophage function., 2015, 113(3): 505–512.
[37] Ichiyama K, Chen TT, Wang XH, Yan XW, Kim BS, Tanaka S, Ndiaye-Lobry D, Deng YH, Zou YL, Zheng P, Tian Q, Aifantis I, Wei L, Dong C. The methylcytosine dioxygenase Tet2 promotes DNA demethylation and activation of cytokine gene expression in T cells., 2015, 42(4): 613–626.
[38] Lal G, Bromberg JS. Epigenetic mechanisms of regulation of Foxp3 expression., 2009, 114(18): 3727–3735.
[39] Nakatsukasa H, Oda M, Yin JH, Chikuma S, Ito M, Koga-Iizuka M, Someya K, Kitagawa Y, Ohkura N, Sakaguchi S, Koya I, Sanosaka T, Kohyama J, Tsukada YI, Yamanaka S, Takamura-Enya T, Lu QJ, Yoshimura A. Loss of TET proteins in regulatory T cells promotes abnormal proliferation, Foxp3 destabilization and IL-17 expression., 2019, 31(5): 335–347.
[40] Ravanan P, Srikumar IF, Talwar P. Autophagy: The spotlight for cellular stress responses., 2017, 188: 53–67.
[41] Peng J, Yang Q, Li AF, Li RQ, Wang Z, Liu LS, Ren Z, Zheng XL, Tang XQ, Li GH, Tang ZH, Jiang ZS, Wei DH. Tet methylcytosine dioxygenase 2 inhibits atherosclerosis via upregulation of autophagy in ApoE–/–mice., 2016, 7(47): 76423–76436.
[42] Yang Q, Li XH, Li RQ, Peng J, Wang Z, Jiang ZS, Tang XQ, Peng Z, Wang Y, Wei DH. Low shear stress inhibited endothelial cell autophagy through TET2 downregulation., 2016, 44(7): 2218–2227.
[43] Xu YJ, Zheng L, Hu YW, Wang Q. Pyroptosis and its relationship to atherosclerosis., 2018, 476: 28–37.
[44] Zeng ZL, Chen JJ, Wu P, Liu YM, Zhang TT, Tao J, Wu SY, Xiao JY, Wei DH, Jiang ZS, Wang Z. OxLDL induces vascular endothelial cell pyroptosis through miR-125a-5p/ TET2 pathway., 2019, 234(5): 7475–7491.
[45] Frismantiene A, Philippova M, Erne P, Resink TJ. Smooth muscle cell-driven vascular diseases and molecular mechanisms of VSMC plasticity., 2018, 52: 48–64.
[46] Liu RJ, Jin Y, Tang WH, Qin LF, Zhang XB, Tellides G, Hwa J, Yu J, Martin KA. Ten-eleven translocation-2 (TET2) is a master regulator of smooth muscle cell plasticity., 2013, 128(18): 2047–2057.
[47] Fang K, Zhang KX, Wang J, Fu ZM, Zhao XH. Advances on the profiling of 5-hydroxymethylcytosine.,2016, 38(3): 206–216.方科, 张凯翔, 王建, 付志猛, 赵湘辉. 表观遗传学新标记—5-羟甲基胞嘧啶检测方法的研究进展. 遗传, 2016, 38(3): 206–216.
[48] Yu M, Han DL, Hon GC, He C. Tet-assisted bisulfite sequencing (TAB-seq)., 2018, 1708: 645–663.
[49] Gibas P, Narmontė M, Staševskij Z, Gordevičius J, Klimašauskas S, Kriukienė E. Precise genomic mapping of 5-hydroxymethylcytosinecovalent tether-directed sequencing., 2020, 18(4): e3000684.
[50] Dong CR, Chen JM, Zheng JL, Liang YM, Yu T, Liu YP, Gao F, Long J, Chen HY, Zhu QH, He ZL, Hu SN, He C, Lin J, Tang YD, Zhu HB. 5-Hydroxymethylcytosine signatures in circulating cell-free DNA as diagnostic and predictive biomarkers for coronary artery disease., 2020, 12(1): 17.
[51] Jiang D, Wang Y, Chang GL, Duan Q, You LN, Sun M, Hu CX, Gao L, Wu SY, Tao HM, Lu K, Zhang DY. DNA hydroxymethylation combined with carotid plaques as a novel biomarker for coronary atherosclerosis., 2019, 11(10): 3170–3181.
[52] Cang S, Lu Q, Ma Y, Liu D. Clinical advances in hypomethylating agents targeting epigenetic pathways., 2010, 10(5): 539–545.
[53] Zwergel C, Fioravanti R, Stazi G, Sarno F, Battistelli C, Romanelli A, Nebbioso A, Mendes E, Paulo A, Strippoli R, Tripodi M, Pechalrieu D, Arimondo PB, De Luca T, Del Bufalo D, Trisciuoglio D, Altucci L, Valente S, Mai A. Novel quinoline compounds active in cancer cells through coupled DNA methyltransferase inhibition and degradation., 2020, 12(2): 447.
[54] Kao YH, Cheng CC, Chen YC, Chung CC, Lee TI, Chen SA, Chen YJ. Hydralazine-induced promoter demethylationenhances sarcoplasmic reticulum Ca2+-ATPase and calcium homeostasis in cardiac myocytes., 2011, 91(9): 1291–1297.
[55] Jiang QX, Yuan H, Xing XW, Liu JJ, Huang ZJ, Du X. Methylation of adrenergic β1 receptor is a potential epigenetic mechanism controlling antihypertensive response to metoprolol., 2011, 48(5): 301–307.
The role of DNA hydroxymethylation in the regulation of atherosclerosis
Yingchu Hu1,2, Haochang Hu1, Shaoyi Lin1, Xiaomin Chen1,2
As an epigenetic modification, DNA hydroxymethylation plays a significant role in regulating gene expression. In recent years, there has been increasing evidence that suggests abnormal changes of 5-hydroxymethylcytosine (5hmC) and ten-eleven translocation (TET) family proteins in cardiovascular diseases, indicating cardiovascular diseases are closely connected with DNA hydroxymethylation. The level of DNA hydroxymethylation is affected by some common risk factors of atherosclerosis, such as aging, gender, hypertension and smoking. It is also related to the immune and inflammatory reaction involved in the process of atherosclerosis as well as the function of endothelial cells and vascular smooth muscle cells. In this review, we summarize the mechanism and research status of DNA hydroxymethylation and TET family proteins towards atherosclerosis, aiming to provide a reference for the development, diagnosis and treatment of atherosclerosis.
DNA hydroxymethylation; atherosclerosis; 5hmC; TET; epigenetics
2020-04-23;
2020-05-28
浙江省自然科学基金项目(编号:LY19H020003)和宁波市自然科学基金项目(编号:2017A610209)资助[Supported by the Natural Science Foundation of Zhejiang Province (No. LY19H020003), and the Natural Science Foundation of Ningbo City (No. 2017A610209)]
胡颖楚,在读硕士研究生,专业方向:心血管内科。E-mail: lcyxhyc@163.com
陈晓敏,硕士,主任医师,研究方向:心血管内科。E-mail: cxmdoctor@126.com
10.16288/j.yczz.20-056
2020/5/8 9:58:23
URI: http://kns.cnki.net/kcms/detail/11.1913.R.20200507.1717.004.html
(责任编委: 刘峰)
