Cross-talk between microRNAs, long non-coding RNAs and p21Cip1 in glioma: diagnostic, prognostic and therapeutic roles
2020-07-20GiorgioSantoniMariaBeatriceMorelliMassimoNabissiFedericaMaggiOlivieroMarinelliMatteoSantoniConsueloAmantini
Giorgio Santoni, Maria Beatrice Morelli, Massimo Nabissi, Federica Maggi,2, Oliviero Marinelli, Matteo Santoni, Consuelo Amantini
1School of Pharmacy, University of Camerino, Camerino 62032, Italy.
2Department of Molecular Medicine, University of Rome Sapienza, Rome 00161, Italy.
3Medical Oncology Unit, Hospital of Macerata, Macerata 62100, Italy.
4School of Biosciences and Veterinary Medicine, University of Camerino, Camerino 62032, Italy.
Abstract Glioblastoma multiforme is considered one of the most common malignant primary intracranial tumors.Despite treatment with a combination of surgery, chemotherapy and radiotherapy, patients with glioblastoma multiform have poor prognosis.It has been widely accepted that the occurrence, progression, and even recurrence of glioblastoma multiforme strictly depends on the presence of glioma cancer stem cells.The presence of glioma stem cells reduces the efficacy of standard therapies, thus increasing the imperative to identify new targets and therapeutic strategies in glioblastoma patients.In this regard, the p21Cip1 pathway has been found to play an important role in the maintenance of the glioma stem cells.It has been shown that this pathway regulates cancer stem cell pool by preventing hyperproliferation and exhaustion.MicroRNAs, endogenous small non-coding RNAs,and long non-coding RNAs, regulate post-transcription gene expression.These are not only altered in glioma, but also in other cancer types, and are involved in tumor development and progression.Notably, they have also been shown to modulate the expression of proteins in the p21Cip1 signaling pathway.This review highlights the extent and complexity of cross-talk between microRNAs, long non-coding RNAs and the p21Cip1 pathway, and demonstrates how such interplay orchestrates the regulation of protein expression and functions in glioma and glioma stem cells.
Keywords: Glioma, microRNA, long non-coding RNA, p21Cip1, glioma stem cells
INTRODUCTION
According to the World Health Organization (WHO) classification, Glioblastoma multiforme (GBM)is a grade IV malignant glial tumor which displays astrocytic differentiation.It is considered one of the most common malignant primary intracranial tumors[1].Despite treatment with a combination of surgery, chemotherapy and radiotherapy, GBM patients have poor prognosis[2].In 1940, the terms primary glioblastoma (pGBM) and secondary glioblastoma (sGBM) were first used by Scherer to distinguish between rapidly progressingde novotumors and tumors derived from a pre-existing astrocytoma[3].According to Scherer[3], pGBM and sGBM display the same histomorphological hallmarks, while differing in biological properties such as type of evolution, clinical manifestation and progression.The differences among primary and secondary GBM arise at the genotypic and epigenetic levels.It is currently widely accepted that the occurrence, progression and even recurrence of both pGBM and sGBM, depend on the accumulation of mutations in neural stem cells, which undergo transformation into glioma cancer stem cells (GSCs).GSCs are characterized by self-renewal and asymmetric cell division, thus allowing the production of proliferating progenitor cells with stem cell features and differentiated cancer cells[4].Importantly, GSCs are more resistant to radio- and chemotherapy respect to the proliferative progenitors present in the tumor.The p21Cip1pathway plays an important role in the maintenance of the GSC pool because it induces a quiescent state that prevents hyperproliferation and exhaustion.p21Cip1is involved in cell cycle arrest which occurs in response to DNA damage.It stimulates DNA repair, thus reducing genetic instability.Thus, p21Cip1is fundamental in maintaining the GSC pool and its genomic integrity[5].
As the presence of GSCs reduces the efficacy of standard therapy, there is an increased imperative to identify new targets and therapeutic strategies in GBM patients.
P21CIP1: P53-DEPENDENT AND INDEPENDENT REGULATION
The proteins, p21Cip1, p27KIP1and p57KIP2, belong to the Cip/Kip family of cyclin dependent kinase inhibitors(CKIs) and are found in mammals.Although p21Cip1and p27KIP1were initially classified as tumor suppressor proteins due to their involvement in the p53-dependent cell cycle arrest, it is now evident that their roles are more complex.Dysregulation of Cip/Kip protein may provoke specific defects in its tumor suppressor activity, while its oncogenic functions may be preserved, thus supporting cancer development.p21Cip1acts as a tumor suppressor through binding and inhibiting CDK/cyclin complexes and the proliferating cell nuclear antigen (PCNA), thus blocking cell proliferation[6].However, it has been shown that p21Cip1also displays additional functions such as stem cell pool preservation, regulation of cell migration and apoptosis[7,8].While some cancer types do not express p21Cip1, its over-expression or cytoplasmic localization has been associated with poor prognosis in several malignant tumors[8][Figure 1].
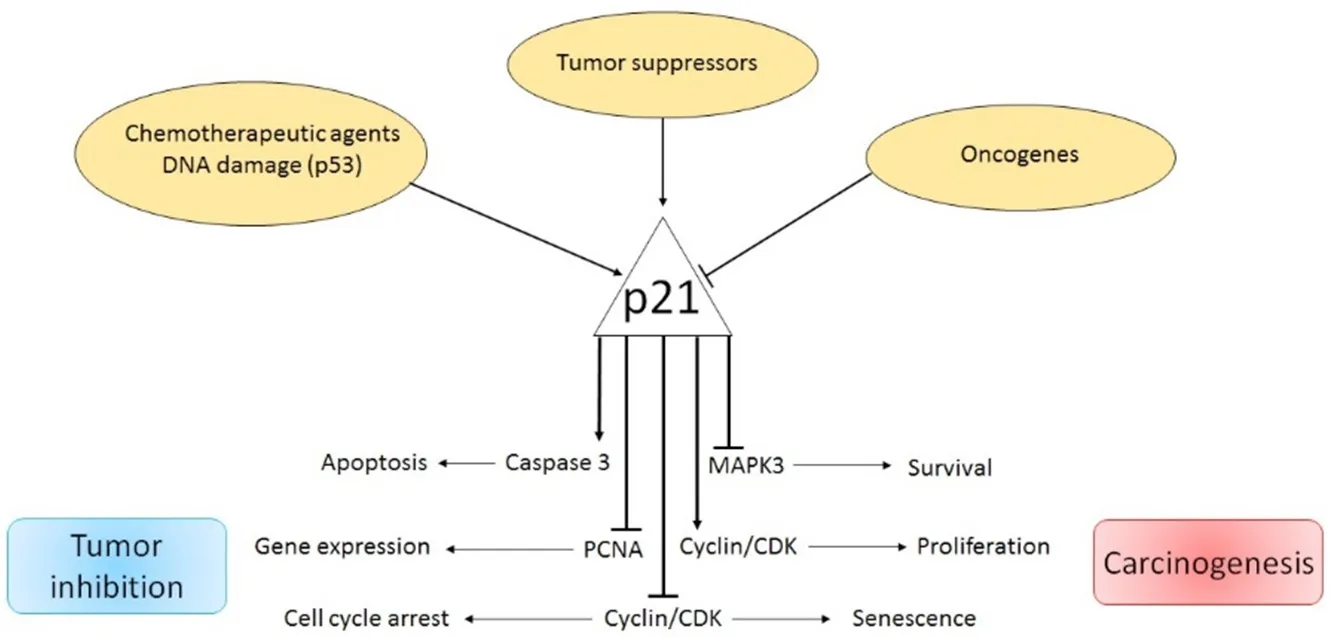
Figure 1.Dual role of p21Cip1 pathway in cancer
p21, a potent cyclin-dependent kinase (CDK) inhibitor, also known as p21Cip1/Waf1/CDKN1A, is a small protein consisting of 165 amino acids.p21 is able to arrest the cell cycle in the G1/S and G2/M phases by inhibiting CDK4, 6/cyclin-D and CDK2/cyclin-E, thus controlling E2F activity[8,9].The principal transcription regulator of p21Cip1is p53.In fact, DNA damage and oxidative stress enhance p53 activity and trigger p21Cip1expression[10].In addition, under the influence of certain factors, the p21Cip1-dependent cell cycle arrest can be also regulated in a p53-independent manner.As such, p21Cip1can be transactivated by the BRCA1 variants in a p53-dependent and independent manner[11].Additionally, transforming growth factor β (TGFβ) and mitogen activated protein kinase control p21Cip1expression by phosphorylation of SMAD1 and SMAD3, while c-Myc over-expression abrogates TGFβ-mediated p21Cip1/SMAD binding[12].The E3 ubiquitin ligase, Makorin Ring Finger Protein-1 [a protein involved in cell cycle regulation] reduces p21Cip1levels by p53 ubiquitination and direct p21Cip1polyubiquitination[13].Contrarily, Double homeobox4,which enhances p21Cip1promoter activity via the transcription factor Sp1, increases p21Cip1expression levels[14].In addition, promoter activity and p21Cip1transcription can be stimulated in a p53-independentpathway via over-expression of β1-integrin receptors, and engagement of the transcription factor Sp1 and the P300 co-activator p21Cip1[15].Down-regulation of p21Cip1mRNA expression can also be induced by c-Myc through interactions with the initiator binding zinc finger transcription factor, MIZ-1[16].Similarly, the promyelotic zinc finger can bind to the p21Cip1promoter, thus blocking the Sp1/p21Cip1promoter interaction and reducing p21Cip1expression[17].The liver kinase B1 is a serine/threonine kinase that activates AMPK and regulates cell growth and apoptosis by triggering a p53-dependent p21Cip1increase[18,19].Enhanced p21Cip1levels, induced by increased p53 and Sp1/DNA binding activity, have been also found in human fibroblasts during replicative senescence[20-22].Moreover, the alkylating agent Temozolomide (TMZ) induces G2-M cell cycle arrest and senescence through the Mre11-Rad50-Nbs1 (MRN) complex and activation of the ATR/CHK1 axis.TMZ-induced senescence requires a functional p53, a functional NF-κB pathway, as well as a sustained p21Cip1 induction.Upon TMZ exposure, as a consequence of the E2F1/DP1 complex disruption,a strong repression of the mismatch repair proteins MSH2, MSH6, EXO1 and RAD51 was observed[23].
The Wnt signaling pathway is a critical regulator of cancer stem cell (CSC) features and has been associated with poor prognosis.Its downregulation is responsible for diminished numbers of CD133+CD44+CSCs [in this regard, recent studies on colon cancer have demonstrated that the knock-down of zeste homologue 2(EZH2), a key component of the Polycomb-Repressive Complex 2, is involved in maintaining the transcriptional repressive state of cycle arrest and apoptosis of CSC-like cells[24].EZH2 expression positively correlates with levels of Wnt/β-catenin pathway target genes.EZH2 is essential for the maintenance of CSC-like cell properties] [Figure 2][25].Several findings demonstrate that EZH2 knockdown inactivates the Wnt/β-catenin pathway by increasing p21Cip1expression, resulting in G1/S-phase arrest[25].EZH2 knockdown in CD133+/CD44+SW480 cells has been shown to induce the increase of p21Cip1with consequent reduction of β-catenin, vimentin and c-Myc expression.Moreover, the inhibition of EZH2 via a specific inhibitor promotes enhancement of p21Cip1expression and inactivation of the Wnt/β-catenin pathway[25].
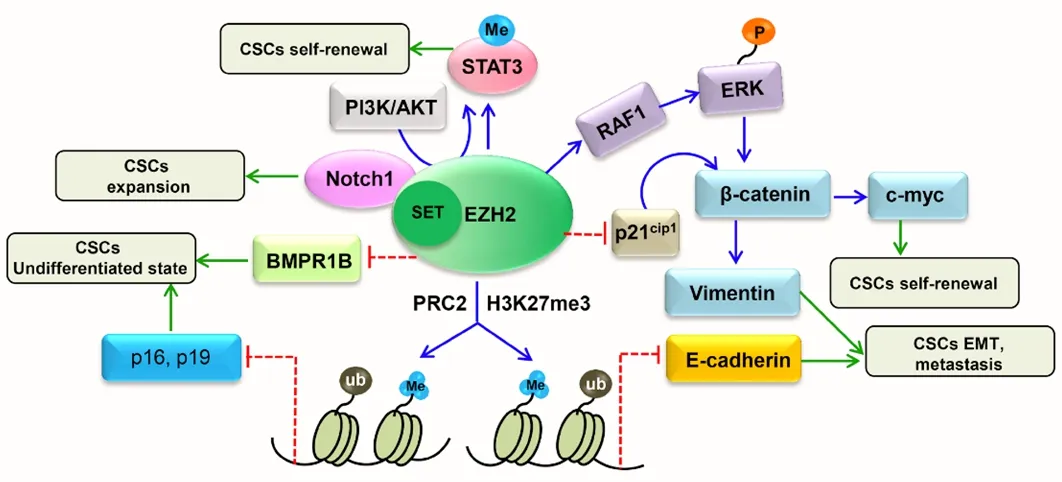
Figure 2.EZH2/WNT/p21Cip1 pathway in cancer stem cells[24]
Interestingly, the role of EZH2 in chemotherapy drug resistance has also been investigated.A decrease in EZH2 expression was demonstrated to promote apoptosis, cell cycle arrest at the G1/S phase, and reduce expression of multi-drug resistant proteins in TMZ-resistant GBM cells[26].GSC maintenance involves the cell-division cycle protein 20 (CDC20), an activator of the E3 ubiquitination ligase complex.SilencingCDC20 expression attenuates GSC proliferation, self-renewal andin vivotumor growth by inducing apoptosis and cell cycle inhibition.CDC20 maintains the GSC phenotype through p21Cip1degradation.CDC20 inhibition stabilizes p21Cip1and represses Survivin, CDC25C and c-Myc survival gene expression[27].
In addition, the roles of p21Cip1and p27KIP1in GSCs treated with camptothecin [CPT, a specific inhibitor of the DNA topoisomerase I that induces DNA double-strand breaks in GSCs carrying the homozygous CDKN2A/ARF deletion] have recently been investigated[28].The results showed that both p21Cip1and p27KIP1proteins block the cell cycle in both unstressed conditions and in response to genotoxic stress.Cip/Kips expression was upregulated after CPT treatment, with a peculiarly nuclear localization.CKIs protect cancer cells against CPT damage by stopping cell cycle progression.Notably, cells become more susceptible to DNA damage in the absence of p21Cip1and p27KIP1proteins, leading to impaired cell cycle blockage under genotoxic stress.Overall, the existing literature demonstrates that p21Cip1and p27KIP1may act both as tumor suppressors by reducing cell proliferation, and as oncogenes, by increasing cellular resistance in response to DNA damage.A deeper understanding of Cip/Kip functions may be relevant and useful to improve understanding on the mechanisms underlying the acquisition of chemoresistance in cancer[28].
Finally, p21Cip1is one of the major regulators of cell cycle and has been previously linked to apoptosis resistance in glioma cells[29].O(6)-Methylguanine-DNA methyltransferase (MGMT) plays a major role in the resistance to alkylating agents in gliomas[30].Happoldet al.[31]demonstrated that in TMZ-resistant glioma cell line, constitutively expressing MGMT, but not in glioma cell lines negative for MGMT, a strong up-regulation of MGMT levels and elevated p21Cip1mRNA levels and slower cell cycle progression[31];however, silencing p21Cip1silencing in resistantvs.normal glioma cells does not evidenced major changes in cell cycle distribution.Mostofaet al.[32]clarified the role for p21Cip1in TMZ-resistance in glioma cells-the p21Cip1protein sequestrates PCNA by binding to it during the cell cycle, thus attenuating DNA replication.In human GBM, MGMT harbors a PCNA-interacting protein motif (PIP box), thus associating with PCNA and in turn, p21Cip1.PCNA is strictly controlled by p21Cip1, a strong binder and sequestrator of the former.Alkylating DNA damage induces MGMT and PCNA colocalization, and this occurs mainly in glioma cells deficient in p21Cip1, as p21Cip1expression directly correlates with higher MGMT mRNA and protein levels,higher MGMT activity and greater resistance to alkylating agents.p21Cip1strongly disrupts MGMT-PCNA complexes within glioma cells.MGMT proteins are downregulated at mid to late S-phase and specifically depredated by the PCNA-dependent ubiquitin-ligase, CRL4Cdt2.
As the p21Cip1protein harbors binding domains for CDKs and PCNA, it serves as an upregulator of MGMT expression, enhancing its transcription and expression, thus providing glioma resistance to alkylating anticancer drugs.
P21CIP1 AND MIRNA IN GLIOMA
MicroRNAs (miRs) are endogenous small non-coding RNAs which regulate gene expression posttranscriptionally.They are commonly deregulated in different types of cancer, including gliomas.In cancer cells, miR regulate cell viability, proliferation, differentiation, migration, invasion, apoptosis,chemoresistance and radioresistance.They can function as oncogenes by promoting cell growth or as antioncogenes by suppressing tumor growth[33].
Recent reports indicate a strict relationship between the p21Cip1signaling pathway and miRs [Table 1].The expression of miRs is significantly upregulated in gliomas as compared to that of adjacent nontumor tissues or normal brain tissue.The expression of miR-10b, -17-92, -92b, -93, -149, -193b, 6798-3p was elevated in glioma tissues as compared to normal controls and significantly increased with tumor grade progression.It has been demonstrated that survival of GBM patients expressing high levels of miR-10 is lower in comparison with patients with low miR-10 levels indicating that miR-10 may contribute to glioma growthin vivo[34].miR-10b inhibition enhances the expression of direct miR-10b targets, namely BCL2L11/Bim, TFAP2C/AP-2γ, CDKN1A/p21, and CDKN2A/p16, thereby reducing glioma cell growth by cell-cycle arrest and apoptosis.In cells expressing high levels of p21Cip1, miR-10 represses E2F1-mediated transcription, leading to down-regulation of miR-15/16 and E2F1 target genes, thus delaying progression through to S-phase of the cell cycle.Consequently, miR-15/16 activities are reduced through the repression of many of its targets such as FBXW7, the ubiquitin ligase that destabilizes Cyclin E.However, miR-10b inhibition induces a weaker E2F1 response in GBM cells that express low levels of p21Cip1[35].Using the clustered regularly interspaced short palindromic repeats-cas9 system, it has also been demonstrated that ablation of the miR-10b gene, through alterations in the expression of several targets including p21Cip1,strongly reduces glioma cell viability in bothin vitroandin vivoexperimental models[36].This suggests miR-10b gene editing to be a promising therapeutic approach for permanent elimination of essential regulators of tumor survival.

Table 1.miRs targeting p21Cip1 in glioma
In addition, the miR-17-92 locus has been found to be amplified in GBM specimens.Inhibition of miR-17-92 activities enhances apoptosis and reduces cell proliferation of GBM spheroids.miR-17-92 inhibition has also been associated with increased expression of CTGF, a direct target of miR-17-92 in GBM spheroids, as well as CDKN1A, E2F1, PTEN[37].
Several target of miRs are represented by members of the TGF/SMAD pathways.In GBM tissues overexpressing miR-92b, the expression of SMAD3 was found to be reduced when compared with that of normal brain tissues.miR-92b directly affects SMAD3 expression by targeting the 3’-untranslated region.Silencing miR-92b inhibits GBM viability through upregulation of the TGF/SMAD3/p21Cip1signaling pathway.Furthermore,in vivotreatment with miR-92b inhibitors was shown to reduce tumor growth,demonstrating the ability of miR-92b to act as an oncogene by promoting GBM cell proliferation.Therefore,miR-92b may be a future potential target for the development of miR-based therapies[38].miR-193b has also been shown to be upregulated in glioma patients, while its overexpression has been associated with poor prognosis.Downregulation of miR-193b correlates with decreased cell growth.SMAD3 is a direct target of miR-193b, and downregulation of SMAD3 attenuates miR-193b suppression of glioma proliferation.Thus,miR-193b regulates cell growth through the TGF-β pathway by modulating SMAD3[39].
Upregulation of miR-93 has also been demonstrated to be associated with advanced malignancy as it promotes the proliferation, migration and invasion of glioma cells.miR-93 regulates the cell cycle by controlling the p21Cip1, p27KIP1, p53 and cyclin D1 expression.Since p21Cip1is a direct target of miR-93,p21Cip1knockout attenuates the suppressive effects of miR-93 on cell cycle progression and colony formation.In addition, the chemosensitization of GBM cells to temozolomide is markedly increased when miR-93 is inhibited[40].Overexpression of miR-149 in gliomas augments pro-survival activity, inhibits apoptosis and induces xenografted tumor growthin vivo.Given that caspase-2 is a functional target of miR-149, its expression is inversely associated with miR-149in vitro.miR-149 promotes cell survival in U87 and A172 glioma cell lines and targets caspase-2, through p53 and p21Cip1inactivation[41].
Several miRs have be found to be down-regulated in glioma patients, thus functioning as tumor suppressor genes (e.g., miR-34a, -128, -184, -223 -329 and -656).miR-34a maps to chromosome 1p36.23, a region often deleted in GBM.For this reason, the expression of miR-34a is lower in GBM samples as compared with that of normal brain tissue.In glioma patients, an miR-34a deletion is accompanied by the amplification of epidermal growth factor receptor (EGFR).Notably, mean survival time is shorter in GBM patients with EGFR amplification and miR-34a deletion[42].Moreover, enforced expression of miR-34a in GBM cells decreases migration and levels of cyclin-A1, -B1, -D1, and -D3, as well as cyclin-dependent kinases, while increasing the expression of cyclin kinase inhibitor proteins such as p21 or p27KIP1.Inin vivoxenograftmouse model, the injection of U251 cells overexpressing miR-34a induced the development of smaller tumors compared with tumors derived from wild-type U251 cells.Since miR-34a targets Yin Yang-1, a transcription factor that stimulates the expression of EGFR, the expression of EGFR is reduced in cells overexpressing miR-34a.miR-34a act as a tumor suppressor by inhibiting the growth of GBM cellsin vitroandin vivo[42].
miR-128 expression reduced glioma cell proliferationin vitroandin vivoin a glioma xenograft growth model.It reduced Bmi-1 oncogene expression, by direct regulation of the Bmi-1 mRNA 3’-untranslated region, through a miR-128 binding site.Relative to normal brain tissues, Bmi-1 is upregulated and miR-128 is down-regulated in glioma samples.Bmi-1 induces the silencing of several genes through epigeneticchromatin modifications.It is responsible for decreasing histone methylation [H3K27me(3)] and Akt phosphorylation, while upregulating p21Cip1levels.Bmi-1 also promotes self-renewal of stem cells, while miR-128 [by down-regulating Bmi-1 levels] inhibits self-renewal of glioma[43].miR-184 levels are reduced in aggressive human tumor cells.Thus, a potential therapeutic strategy which targets miR-184 may prove useful in reducing cancer aggressiveness.miR-184 inhibits the proliferation and invasion of the U87MG cell line.It arrests the cell cycle and prevents adhesion by upregulating the expression of p53 and p21Cip1,increasing caspase-3/8 activity, suppressing SND1, MMP-2/9, CD44 expression, and the activity of the AKT/NF-κB pathway[44].Another miR involved in glioma development is miR-223.It represses nuclear factor I-A (NFIA) expression, a key regulator of gliogenesis.miR-223 and NFIA expression negatively correlate in human GBM tumors.The miR-223/NFIA axis suppresses tumorigenesis in human glioma cells.The NFIA factor directly represses p21Cip1, and is required for tumorigenesis in a mouse neural stem cell model of glioma[45].
miR-329, which is located on 14q32.31, is down-regulated in glioma and it is able to target E2F1.Its overexpression blocks G1/S transition in LN18 and T98G cell lines and suppresses cell proliferation as well as colony formation.It also decreases the phosphorylation of Akt and cyclin D1, inducing p21Cip1upregulation and suppression of cell growth through the inhibition of the E2F1-mediated Akt pathway[46].
miR-656 was found to be downregulated in glioma.In human glioma tissues, expression of BMPR1A [a miR-656 target] is negatively correlated with miR-656 levels.miR-656 suppresses glioma cell proliferation,neurosphere formation, migration and invasion with or without exogenous BMP-2[47].Knockdown of BMPR1A diminished the antiproliferative effect of miR-656in vitro.Moreover, the canonical BMP/SMAD pathways were shown to be inhibited by miR-656 overexpression.Several molecules, including cyclin B,cyclin D1, matrix metalloproteinase-9, p21Cip1and p27KIP1are involved in miR-656 functions within glioma cells.Ectopic expression of miR-656 reduced tumor size and prolonged the survival of mice, regardless of whether treatment with BMP-2 was administered[47].
Some miRs, even if down-regulated in gliomas, function as tumor promoters.For example, an important role has been described for miR-454, which is down-regulated in GBM primary tumors and cell lines.Overexpression of miR-454 in GBM cells resulted in the arrest at the G0/G1 phase, resulting in the inhibition of cell proliferation.3-phosphoinositide-dependent protein kinase-1 (PDK1), one of miR-454 targets, was found to increase miR-454 levels, decrease PDK1 expression, down-regulate Cyclin D1 and upregulate p-Rb and p21Cip1[48].
In addition, arsenic resistance protein 2 (Ars2), a component of the nuclear RNA cap-binding complex,that is involved in miR biogenesis, proliferation and tumorigenicity, was found to be overexpressed in glioma cell lines[49].It is also associated with poorer overall survival in GBM patients[49].In fact, not only several miRs have been found to be associated with pathological grade, tumor development and stemness in glioma[50,51], but also it has been demonstrated that overexpression of Ars2 stimulates glioma cell proliferation and colony formation and colony formation.Knockdown of Ars2 reduces miR-6798-3p expression, causing p53 and p21Cip1upregulation which leads to apoptosis.By using an orthotopic GBM xenograft model, knockdown of Ars2 was found to reduce tumor growth and to extend the survival time of tumor-bearing mice[49].
ROLE OF LONG NON-CODING AND LONG INTERGENIC NON-CODING RNAS IN GLIOMA AND GLIOMA STEM CELLS
Long noncoding RNAs (LncRNAs) represent a class of long transcribed noncoding RNAs (ncRNA)molecules, which are longer than 200 nucleotides.They do not code for proteins and are involved in tumordevelopment and progression.Long intergenic noncoding RNAs (LincRNAs) are long RNA transcripts which control cell differentiation and maintenance of cell identity.All of the above have recently been found to be altered in various cancer types [Table 2][52].LncRNAs have been recognized as regulators involved in different steps of the tumorigenic process.In gliomas, the functions of most LncRNAs are not well known, and the mechanisms controlling the proliferation, invasion, angiogenesis, radiosensitivity or radioresistence, and GBM stemness remain poorly defined[53].While many LncRNAs have been identified,only a few have been functionally described in gliomas Moreover, CSC regulation by LncRNA following radiotherapy and the relationship between LncRNA and tumor spreading and radioresistance have been reported[54].
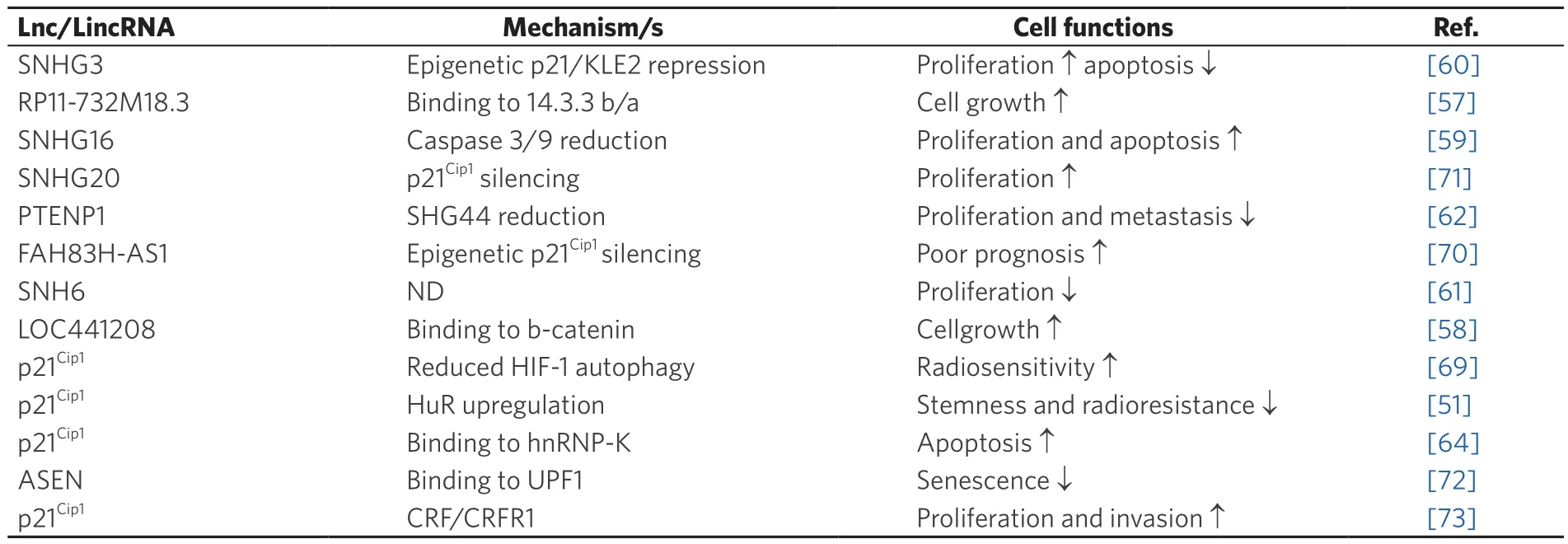
Table 2.Long non-coding RNA and long intergenic non-coding RNA interacting with p21Cip1 pathway in glioma
Recent studies have evidenced that LncRNAs control the transcription of genes involved in DNA Damage Response (DDR), the latter of which is strictly associated with radiosensitivity and repair capacity[55,56].The majority of LncRNAs are overexpressed in tumor cells, hence their inhibition might represent a new therapeutic target for glioma treatment.
Among LncRNA, RP11-732M18.3 is highly overexpressed in glioma cells and functions as an oncogene by interacting with 14-3-3β/α to promote glioma growth[57].
Its overexpression has been associated with the proliferation of glioma cells and tumor growth bothin vitroandin vivo.LncRNA RP11-732M18.3 stimulates the G1/S cell cycle transition and cell proliferation.It has been found that interaction of LncRNA RP11-732M18.3 with 14-3-3β/α increases the degradation of the p21Cip1protein.In fact, by promoting the recruitment of ubiquitin-conjugating enzyme E2 E1 (UBE2E1)to 14-3-3β/α and with the binding of 14-3-3β/α with UBE2E1, LncRNA RP11-732M18.3 stimulates the degradation of p21Cip1[57].Similarly, LOC441204-LncRNA, has been shown to be upregulated in glioma cell lines.It promotes tumor cell growth by stabilizing the β-catenin pathway.LOC441204 can bind to β-catenin and prevent its degradation, thus resulting in downstream p21Cip1repression and cdk4 activation that in turn enhances glioma cell proliferation.The LOC441204 knockdown suppressed tumor cell proliferation of glioma cell lines[58].
The expression of the small nucleolar RNA host gene 16, SNHG16-LncRNA was upregulated in gliomas.Knockdown of SNHG16 is associated with high p21Cip1expression, poor proliferation and increased apoptosis.Lnc-SNHG16 was shown to inhibit p21Cip1expression and caspase 3/9 activation, while increasing cyclinD1/B1 expression[59].The SHNG3 upregulation in glioma promoted cell proliferation,accelerated cell cycle progression and repressed apoptosis.SNHG3 facilitated the malignant progression of glioma through epigenetically repressing KLF2 and p21Cip1via recruitment of the enhancer of zeste homolog 2 to the promoter of KLF2 and p21Cip1[60].Similarly, overexpression of SNHG6 promotes the malignant phenotype, while loss-of-function revealed that the silencing of SNHG6 inhibits glioma cell growth in a p21Cip1-dependent manner[61].
The LncRNA PTEN pseudogene-1 (PTENP1) has been described to be involved in the development and progression of several cancers.However, little is known about the molecular mechanism by which lncRNA PTENP1 affects the development and progression of gliomas.Overexpression of PTENP1 suppressed SHG44 expression as well as U251 cell proliferation, invasion and migration by inducing p21Cip1expression while inhibiting p38 signaling[62].
Among LincRNA, the LincRNA-p21Cip1is an intergenic LncRNA which resides on the chromosome 17,upstream of the p21Cip1gene.It was identified ten years ago as being one of the eleven LincRNAs that are increased in expression in response to p-53 induced DNA damage[63].
LincRNA-p21Cip1is the downstream target of p53 and controls the cell cycle, damage of DNA and its repair process[63].The LincRNA-p21Cip1suppresses the p53-dependent transcription activity and affects p53-repressed genes.It is required to trigger p53-dependent apoptosis of DNA damaged cells through physical association with the ribonucleoprotein K (hnRNP-K)[64].
LincRNA-p21Cip1also regulates the G1/S checkpoint of the cell cycle by modulating the expression of its neighboring gene p21Cip1[65]and mediating radiation-induced apoptosis[66,67].LincRNA-p21Cip1is a potent suppressor of GSC and works by triggering apoptosis through the NOXA pro-apoptotic gene[68].It negatively regulates β-catenin activity in GSCs.Downregulation of LincRNA-p21Cip1in GSCs results in upregulation of Hu antigen R (HuR), and this is caused by miR-146b-5p down-regulation.Moreover,knockout of LincRNA-p21Cip1or HuR was shown to increase GSC β-catenin expression, stemness and GSCs radioresistance[51].Previous findings also showed that LincRNA-p21Cip1is a hypoxia-responsive lncRNA able to regulate the cell cycle and apoptosis.It has also been shown to be responsible for the Warburg effect in cervical cancer.Although hypoxia was found to increase LincRNA-p21Cip1expression in U251MG glioma cell line, its functions in hypoxic glioma remain unknown.Knockdown of LincRNA-p21 induced G2/M phase arrest, promoted apoptosis, decreased cell proliferation and motility, and also reduced autophagy through the HIF-1/Akt/mTOR/P70S6K pathway[69].These findings are suggestive of a mechanism by which LincRNA-p21Cip1enhances the radiosensitivity of hypoxic tumor cells.
The antisense FAM83H RNA1 (FAM83H-AS1) is known to be upregulated in glioma.Silencing of FAM83H-AS1 suppressed glioma proliferation and apoptosis.FAM83H-AS1 inhibits CDKN1A expression by recruiting EZH2 to the promoter of CDKN1A, thereby influencing cell cycle and proliferation[70].
LincRNAs also regulates cellular senescence.Thus, LincRNA-ASEN, which is expressed in prematurely senescent cells, has been shown to repress cellular senescence by reducing p21Cip1expression, both during and post-transcription.The complex formed by the interaction between Linc-ASEN and UPF1 can suppress p21Cip1transcription through the recruitment of Polycomb Repressive Complex 1 and 2 (PRC1 and PRC2) to the p21 locus.This prevents binding of the transcriptional activator p53 to the p21 promoter through histone modification[64].
CLINICAL ROLE OF MIRNA TARGETING P21CIP1 AND LNCRNA-P21CIP1 IN GLIOMAS
The identification of glioma-related biomarkers is an urgent necessity as it would enable the development of new therapeutic approaches, while improving the ability to predict responsiveness to chemo- andradiotherapy.In this regard, several miRs including miR-10b[34]and miR-92b[38]have been suggested as targets for the development of miRNA-based therapies.Inhibition of miR-93 enhanced the chemosensitization of glioma cells to TMZ treatment[40], while miR-17-92 and its target CTGF have been suggested for use in differentiation-promoting therapy in the treatment of GSC[37].Conversely, the upregulation of miR-193b[33]and miR-93 are associated with advanced malignancy[40]and represent negative prognostic factors.Ars-2, which reduces miR-6798 expression, was associated with poor overall survival in glioma patients[49].
Finally, Shenet al.[41]have suggest that the miR effect in cancer may also depend on the p53 status of tumor cells.In glioma cell lines harboring wild-type p53, miR-149 showed pro-survival function, as it targeted caspase-2 via the inactivation of p53/p21Cip1pathways, while functioning as a tumor suppressor in proliferation and invasion of glioma cell lines with p53 mutations.miR-149 also inhibited proliferation and migration of glioma cell lines with p53 loss, suggesting its distinct biological function within both p53 wild-type and p53 mutated glioma cells.
LncRNAs have recently emerged as crucial players in the p21Cip1complex signaling network as they control glioma development and progression[71-73], the activation of GSCs, as well as radioresistance.LncRNAs aberrantly expressed in CSCs in different cancer types such as epatocarcinoma, are active participants in the major signaling pathways governing DDR, DNA repair, apoptosis and epithelial-mesenchymal transition[54,74].It has been also reported that the deregulation of LncRNAs plays a role in cancer recurrence and prognosis[75].With increasing knowledge on the expression profile of LncRNAs in cancers, their potential roles as biomarkers in diagnosis and prognosis have been highlighted.Expression levels of LncRNAs are closely associated with the real tumor status.Additionally, LncRNAs are more sensitive and specific as compared to the other conventional markers.Furthermore, given that LncRNAs are released in body fluids, this suggests their potential to be utilized in a clinical setting as non-invasive biomarkers[75].
In gliomas, several dysregulated LncRNAs are involved in proliferation, radioresistance, metastasis and cancer stem cell properties.A strong correlation between high LncRNA expression and clinical parameters has been reported for LncRNA-SNHG20 in glioma patients.High LncRNA-SNHG20 has been associated with larger tumor size, larger extent of resection, more advanced grade (WHO classification),poorer survival status and a higher incidence of recurrence.High SNHG20 levels are predictive of poor prognosis and represent an independent potential prognostic biomarker for glioma patients[71].Similarly,overexpression of SNHG3, SNHG6 and FAH83H-AS1, which promote the malignant phenotype, represent a negative prognostic factors[60,61,70].
Given the up-regulated expression of LncRNAs in glioma patients, and the role in cancer development and progression, LncRNA inhibition (e.g., RPH, SNHG3, SNHG16, LOC441204)[57-60]would represent a novel diagnostic and therapeutic strategy in gliomas.Moreover, as LincRNA-p21Cip1enhances the radiosensitivity of hypoxic tumor cells, it represents a valuable target for radiation therapy in glioma patients[69].Furthermore, in GSCs, the LncRNA-mediated effects also depend on the epigenetic regulation of genes,particularly via recruitment of the Polycomb repressor complex and by acting as competing endogenous RNAs for miRNAs targeting genes involved in stemness and radioresistance, mainly of the WNT/B-catenin and p21Cip1pathways[76].
Finally, several miRs such as miR26a[77]and LncRNAs such as Lnc-TALC[78]have been found to promote MGMT expression and TMZ resistance in gliomas.However, at present no miRs or LncRNAs targeting p21Ciphave been employed to modulate alkylating agent resistance in gliomas.
CONCLUSION
The functions of the vast majority of miRNAs, LncRNAs and LincRNAs encoded by mammalian genome remain largely unknown.However, recent studies indicate that these transcripts play vital roles not onlyin cellular physiology, but also in cancer pathology.In this regard, alternations in their regulation have been found to be implicated in the development and progression of glioma[53,79,80].Complex cross-talk and interplay between miRNAs, Lnc/LincRNAs and p21Cip1orchestrate the regulation of protein expression and functions in cancer cells.Our understanding of the functional roles and molecular mechanisms of miR and LncRNAs in glioma and GSC is still nascent.Several findings indicate that miRs targeting p21Cip1and more recently LncRNAs signatures correlate with glioma malignancy grade, histological differentiation and prognosis[47,51,70].Further in-depth investigation into miRs, LncRNAs and LincRNAs, their complex mechanisms of regulation, as well as their involvement in glioma genesis, metastatic spread and therapeutic resistance will be necessary in order to develop novel and effective diagnostic and therapeutic strategies against primary and secondary GBM tumors.
DECLARATIONS
Authors’ contributions
Supervised the work and wrote the manuscript: Santoni G
Contributed to the preparation of the subchapters: Morelli MB, Amantini C
Contributed to the preparation of figures and tables: Maggi F, Marinelli O
Provided critical revision of the manuscript: Nabissi M
Collaborated in the Drafting of the introduction and conclusion: Santoni M
Availability of data and materials
Not applicable.
Financial support and sponsorship
This work was supported by PRIN 2017 and Fondazione Umberto Veronesi (Post-doctoral Fellowship 2019 to Morelli MB).
Conflicts of interest
All authors declared that there are no conflicts of interest.
Ethical approval and consent to participate
Not applicable.
Consent for publication
Not applicable.
Copyright
© The Author(s) 2020.
杂志排行

Journal of Cancer Metastasis and Treatment的其它文章
- Magnetic nanotechnologies for early cancer diagnostics with liquid biopsies: a review
- Lobe-specific modulation of B16MET melanoma lung metastases by nephrilin peptide
- Mouse tumor susceptibility genes identify drug combinations for multiple myeloma
- Could zinc dipicolinate be used to “smuggle” zinc into prostate cancer cells?