聚乳酸-b-聚异戊二烯嵌段共聚物的合成及其对聚乳酸/橡胶的增韧研究
2020-07-17夏登周沈显荣高建纲相益信
夏登周,沈显荣,高建纲,相益信
(安徽工程大学 生物与化学工程学院,安徽 芜湖 241000)
聚乳酸(Polylactic Acid,PLA)是一种生物相容性非常好的高分子材料,它在堆肥后可完全降解为CO2和水,在土壤和水质中无塑料残留。且它的原料来源于自然界,可大规模地进行合成,目前在医学领域、纺织包装等行业[1-5]得到了广泛的应用。但是聚乳酸也存在许多不足,比如韧性差、脆性高、热稳定性差等,这些不足给它在取代其他塑料时造成了一定的限制[6-9]。为了改善聚乳酸的缺陷,科学家们进行了大量的改性研究,改性手段也多种多样,主要有共混、复合、共聚、增塑及添加成核剂等以提高PLA的力学性能,扩大其应用范围[9-11]。天然橡胶(Natural Rubber,NR)是目前使用非常广泛的一种天然高分子化合物,它的主要成分是顺1,4-聚异戊二烯,其拥有弹性好、绝缘性高、可塑性强等一系列优点,成为了最受青睐的通用橡胶之一,且天然橡胶属于可再生资源,生物相容性和降解性能都非常优越。在塑料中添加天然橡胶以提高塑料的韧性一直是材料领域的重点研究方向,所以近几年在聚乳酸中添加天然橡胶多有报道[12-14]。然而,由于天然橡胶和聚乳酸二者间的极性差别很大,彼此之间不能很好地融合在一起,产生明显的宏观相分离,从而使材料的力学性能不能达到预期效果,因此调控二者的相容性非常重要。张春梅[15]等在聚乳酸/天然橡胶加入过氧化二异丙苯(DCP)后测试聚乳酸性能,结果表明,DCP使天然橡胶与PLA发生交联,PLA和NR很好地相容在一起,经过测试纯聚乳酸的冲击强度只有2.2 kJ/m2,而改性的聚乳酸提高到了7.2 kJ/m2,同时断裂伸长率也提升到了原来的2倍;HG.Ock[16]等使用有机黏土作相容剂将其加入PLA与NR的混合物中,研究表明,天然橡胶颗粒很好地分散在聚乳酸相中,从而改性聚乳酸;ML.Robertson[17]通过加入PI-b-PLA嵌段共聚物作相容剂,使聚合后的大豆油与聚乳酸融合后增强韧性,这种方式虽然成功地改性了聚乳酸,但是该嵌段共聚物的合成条件比较苛刻,反应要求的温度很高,链段长度不能精确控制。
故研究在此基础上,利用RAFT活性自由基聚合方法,调控PLA-b-PI嵌段聚合物结构,合成了一系列分子链段长度不同的嵌段共聚物,并将其添加进聚乳酸和天然橡胶的混合体系中,探究其对天然橡胶与聚乳酸混合体系增容增韧效果的影响。
1 实验部分
1.1 主要试剂
DL-丙交酯(AR)(郑州阿尔法化工有限公司);甲苯(AR)、二氯甲烷(AR),使用前经分子筛干燥(国药集团化学试剂有限公司);苯甲醇(AR)、辛酸亚锡(AR)、苯并噻唑(AR)、1-(3-二甲氨基丙基)-3-乙基碳二亚胺盐酸盐(简称EDC,AR)、4-二氨基吡啶(简称DMAP,AR)(上海泰坦科技股份有限公司);异戊二烯(简称IP,AR),使用前过碱性氧化铝层析柱除去阻聚剂(上海阿拉丁生化科技股份有限公司);2-(十二烷基三硫代碳酸酯基)-2-甲基丙酸(简称CTA,AR),参考文献[18]合成;聚乳酸2003D,用于共混(购于美国Natureworks公司);天然橡胶,用于共混(海南天然橡胶产业集团股份有限公司)。
1.2 样品制备
(1)丙交酯开环。称取4.844 g(33.64 mmol)丙交酯于100 mL圆底烧瓶中,加入50 mL甲苯减压共沸除去多余的水分至剩余少量甲苯作溶剂。往反应体系里通15 min氮气以排除氧气,后加入苯甲醇0.106 g(0.96 mmol)、辛酸亚锡0.038 8 g(0.096 mmol),三者的摩尔比控制在35∶1∶0.1。再持续通15 min氮气后密封,在130 ℃油浴锅里搅拌24 h,所得产物用少量二氯甲烷溶解稀释后,逐滴滴入到乙醇中沉淀。然后收集沉淀物置于真空干燥箱中30 ℃真空干燥20 h,第二天取出后再重复溶解沉淀干燥两次得聚乳酸。
(2)PLA酯化。将上一步合成的PLA、4-二氨基吡啶、1-(3-二甲氨基丙基)-3-乙基碳二亚胺盐酸盐和2-(十二烷基三硫代碳酸酯基)-2-甲基丙酸(摩尔比=1∶1∶5∶5)依次加入到单口烧瓶中,然后加入50 mL干燥过后的二氯甲烷作溶剂,再在25 ℃水浴锅中持续搅拌48 h。反应结束后减压旋蒸,除去部分二氯甲烷,将产物浓缩,再在甲醇中沉淀干燥3次,得到酯化后的PLA-CTA,该产物用作下一步RAFT聚合的大分子链转移剂。
(3)聚乳酸-b-聚异戊二烯的合成。在耐压瓶中加入异戊二烯、PLA-CTA、过氧化二叔丁基、苯并噻唑(4种物质摩尔比为147∶1∶0.3∶2.53),再加入甲苯作溶剂,通20 min氮气后密封,125 ℃油浴锅里搅拌24 h,再逐滴滴入甲醇中沉淀,30 ℃真空干燥20 h,再用二氯甲烷重复溶解干燥两次得最终产物。合成路线图如图1所示。
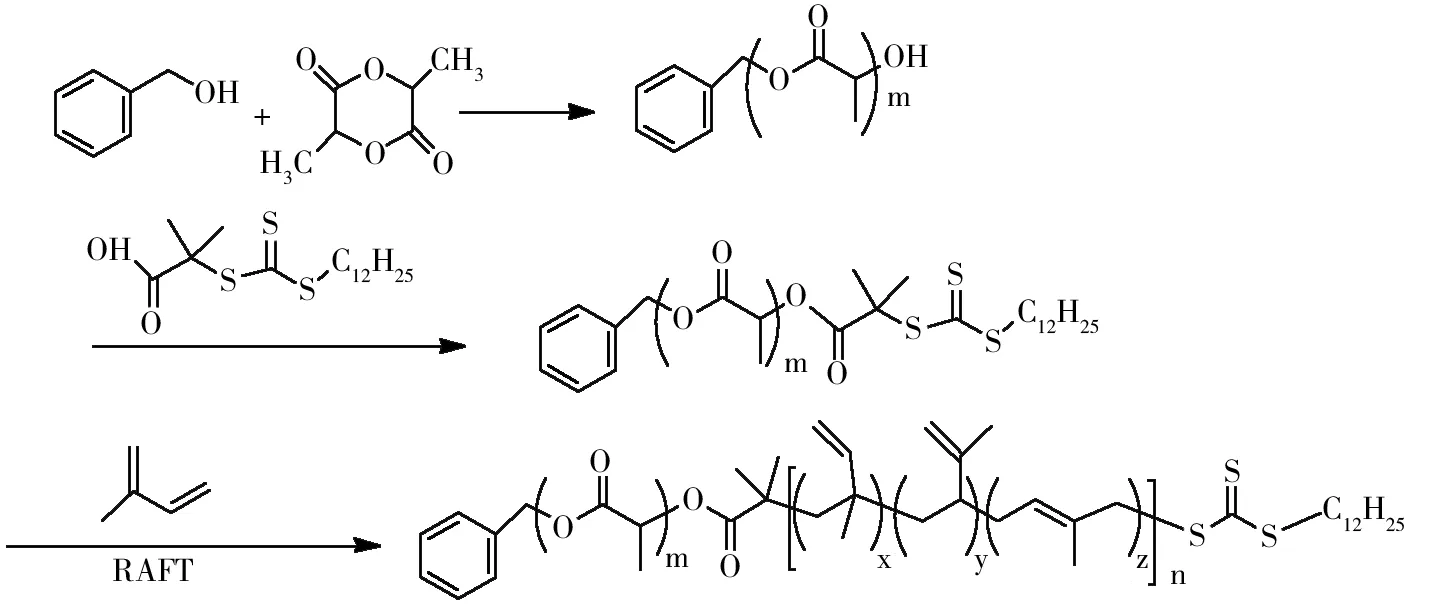
图1 PLA-b-PI合成路线图
1.3 样品的应用
将合成的样品与聚乳酸料粒(2003D)和天然橡胶共混,共混物样品使用密炼机制备,共混前PLA料粒在70 ℃烘箱中干燥12 h。密炼机的温度调成200 ℃,转速设置成60 r/min。所称取的PLA/NR质量比始终保持为9∶1,合成的嵌段共聚物加入量分别占聚乳酸和天然橡胶二者共混物总质量的1%、3%和10%,分别以PLA/NR-1%、PLA/NR-3%、PLA/NR-10%进行表示。密炼结束后趁热将共混后的样品剪成小颗粒备用。
1.4 测试和表征
(1)转化率及产率的测定。采用称重法来确定聚乳酸的产率以及异戊二烯的实际转化率。
(2)分子量的测定。使用凝胶渗透色谱仪(GPC)测定嵌段共聚物的相对分子质量以及它的分子量分布指数,所用仪器型号为Waters 1525。测定条件:柱温设置为40 ℃,四氢呋喃(THF,色谱级)为淋洗液,流速设定为0.5 mL/min。测之前使用聚甲基丙烯酸甲酯(PMMA)标样对仪器进行校准。
(3)核磁氢谱分析。使用400 MHz Ascend 400核磁共振波谱仪确立聚合物结构,各结构基团的比例。溶剂为氘代氯仿。
(4)差示扫描量热分析。采用DSC-100型差示扫描量热分析仪(DSC)分析每个试样的玻璃化转变温度(Tg)、结晶温度(Tc)、熔融温度(Tm)、结晶焓(ΔHc)和熔融焓(ΔHm)。测试时通氮气,升温速度设为10 ℃/min,温度范围为25~200 ℃。在200 ℃保温5 min消除热历史,取二次升温数据。
(5)形貌表征。采用日本JROL公司JSM7800F型扫描电子显微镜(SEM)观察样品经液氮冷却脆断后的断裂面形貌。加速电压为10 KV,测试前样品表面喷金处理。
(6)力学性能测试。采用万能材料试验机测试PLA/NR共混物的拉伸强度、断裂伸长率以及冲击强度,拉伸速度设置为20 mm/min,每组试样测试5个取平均值。冲击测试选择简支梁缺口冲击强度,缺口深度为2 mm,同样每组测5个取平均值。
2 结果与讨论
2.1 丙交酯与异戊二烯转化率及其分子量
在相同的聚合温度和聚合时间下,通过控制投料比来调控得到不同分子量的聚合产物,其产率与GPC测试结果如表1所示。
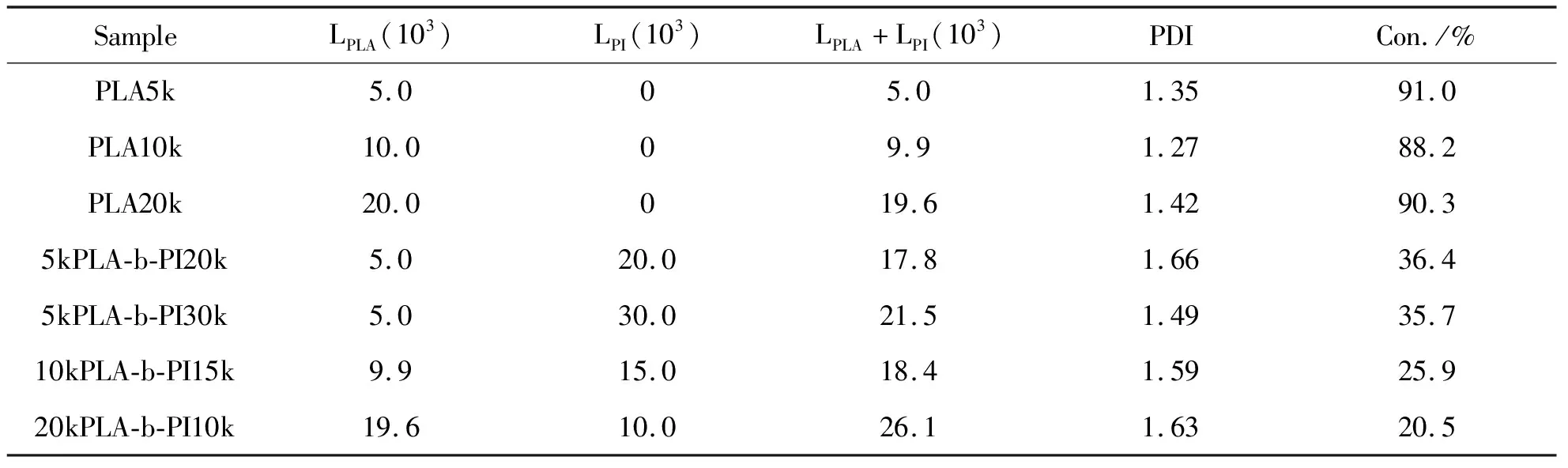
表1 不同分子量的PLA和PLA-b-PI及其分子量分布
注:四种嵌段共聚物投料比nIp∶nCTA∶nI从上到下依次为293.64∶1∶0.3;440.5∶1∶0.3;220.2∶1∶0.3;146.8∶1∶0.3
表1中的丙交酯开环所预设分子量分别为5 k、10 k、20 k,实际制得聚乳酸产率很高,分别为91.0%、88.2%和90.3%;开环聚合的分子量分别为5 000、9 900和19 600,与设计值偏差不大。三者的分子量分布指数分别为1.35、1.27、1.42,相对也比较窄,故可用来作下一步反应的原料。
以得到的聚乳酸链段作基础合成大分子链转移剂PLA-CTA,再分别预设嵌段聚合不同分子量的聚异戊二烯。由GPC测得的实际链段分子量可知,酯化PLA和异戊二烯共聚后,产物的分子量均有不同程度的增长。对比发现,当PLA链段为5 k时,预设的PI分子量为20 k,实际得到的总分子量为17.8 k,接上去的聚异戊二烯链段很大,有12.8 k,异戊二烯转化率相对较高,根据公式计算:

得到该异戊二烯转化率为36.4%;预设的PI分子量为30 k时,实际得到的分子量为21.5 k,接上去的聚异戊二烯链段为16.5 k,异戊二烯转化率计算后为35.7%,该聚合物分子量分布指数为1.49。当PLA链段为10 k时,预设的PI分子量为15 k,实际得到的总分子量为18.4 k,接上去的聚异戊二烯链段变短,为8.4 k,异戊二烯转化率降低,为25.9%,该聚合物分子量分布指数为1.59。当PLA链段为20 k时,预设的PI分子量为10 k,实际得到的PLA-b-PI共聚物总分子量为26.1 k,接上去的聚异戊二烯链段为6.1 k,异戊二烯转化率最低,为20.5%,该聚合物分子量分布在1.63。
通过对比发现,PLA-CTA大分子链转移剂的长度对聚合反应有一定的影响。分子链长度越短,即分子量越小,在该链上发生可逆加成-断裂链转移聚合的效果越好;分子链长度越长,即分子量越大,在链上发生RAFT聚合的效果就越差,异戊二烯转化率也较低。
分子量预设5 k的PLA及其分别预设嵌段15 k、20 k PI的GPC曲线图如图2所示。由图2可以看出,经过嵌段以后的聚合物保留时间变短,即分子量相较于PLA有所增加,且图中的曲线均是单峰,证明了嵌段共聚物没有均聚物的出现,只有共聚物。
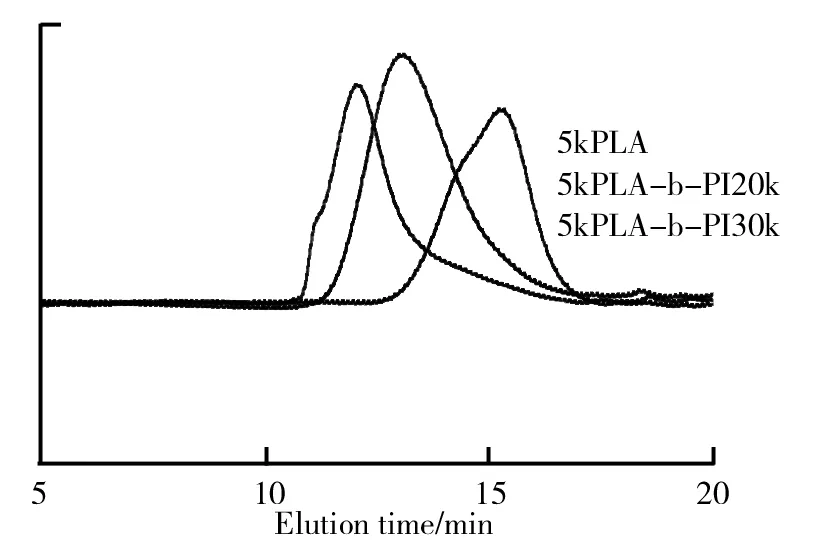
图2 5kPLA及5kPLA-b-PI20k、5kPLA-b-PI30k嵌段共聚物的GPC曲线
2.2 核磁分析
5kPLA-b-PI20k的核磁氢谱图如图3所示。由图3可知,氘代氯仿为7.26 ppm。聚乳酸的上两种不同环境的H,分别为主链碳上的H和侧甲基上的氢,化学位移值为5.21 ppm、1.55 ppm,分别对应谱图中P、O两个位置。
异戊二烯进行RAFT聚合的加成有三种情况:1,2加成、3,4加成和1,4加成。1,2加成的双键碳上有2个H,化学位移值在4.75 ppm,对应d位置,侧甲基上的3个H化学位移值在0.94 ppm,对应k位置;3,4加成的双键碳上也是2个H,化学位移值在4.85 ppm,对应c位置,侧甲基上的3个H化学位移值在1.68 ppm,对应h位置;1,4加成的双键碳上为1个H,化学位移值在5.15 ppm,对应b位置,侧甲基上的3个H化学位移值在1.59 ppm,对应i位置。其余碳上的氢原子化学位移值也可以在谱图上找到对应的位置。通过积分比,即氢原子总数目的比值可计算出PLA链段和PI链段的摩尔比m∶n=0.755∶1.935,由于PLA链段的分子量为5 k,可计算出PI的分子量为12.2 k,聚合物5kPLA-b-PI20k的分子量为17.2 k,与GPC所测得的相对分子质量17.8 k偏差不大,从而验证了所得产物为PLA-b-PI共聚物。另外从图3中也可以得知,异戊二烯进行RAFT聚合后,主要进行的是1,4加成;少部分发生1,2加成和3,4加成。
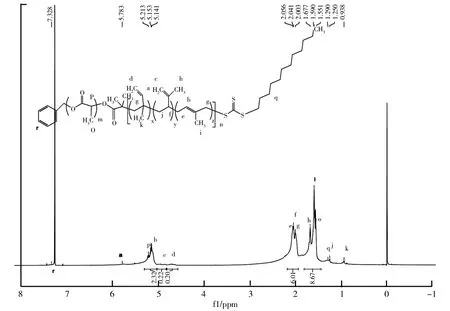
图3 5kPLA-b-PI20k核磁氢谱图
2.3 共混物形貌
(1)PLA和PLA/NR的微观形貌。PLA和PLA/NR的扫描电镜图片如图4所示。由图4可知,纯聚乳酸经液氮脆断后的表面十分光滑,是一种均相体系。而没有加入共聚物的PLA/NR混合物因为并不相容,橡胶颗粒经过脆断被拔出,从而造成了许多孔洞。

图4 液氮脆断后的PLA和PLA/NR扫描电镜图片
(2)加入PLA-b-PI后的PLA/NR微观形貌。PLA/NR加入5kPLA-b-PI20k二嵌段共聚物后的扫描电镜图片如图5所示。聚合物加入量分别占PLA/NR总质量的1%、3%和5%。当加入的PLA-b-PI为1%时,断裂面比未加入聚合物的断面要规整了很多,但是依旧有许多橡胶颗粒被拔出造成的孔洞。当加入的PLA-b-PI为3%时,共混物中只能观察到很少的颗粒分散相,此时天然橡胶已经很好地分散在聚乳酸中,从图5中可以看到橡胶相被拉长变形,不再是大颗粒了。当加入的PLA-b-PI为10%时,从图5中观察不到颗粒分散相了,这是因为大量的PLA-b-PI使聚乳酸和天然橡胶很好地相容在了一起。
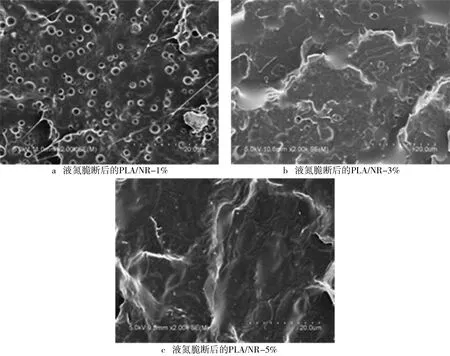
图5 液氮脆断后的PLA/NR-1%、PLA/NR-3%和PLA/NR-5%扫描电镜图片
2.4 热性能分析
由于PLA-b-PI可作相容剂使聚乳酸相和天然橡胶相很好地融合,从而改性聚乳酸,但是在热稳定性上可能会造成影响,故使用差示扫描量热仪对聚乳酸及共混物进行了热性能分析。聚乳酸∶天然橡胶共混物比例依旧为9∶1,密炼选用的样品为5kPLA-b-PI20k链段的,加入量依次为PLA/NR共混物质量的0、1%、3%、10%。密炼后各取5~10 mg,记录下质量后进行DSC测试,所得数据如图6所示。
五种共混物(PLA、PLA/NR、PLA/NR-1%、PLA/NR-3%、PLA/NR-10%)的DSC图谱(向上为放热)如图6所示。从图6中可以看出这五种共混物的玻璃化转变点,向上的结晶放热峰以及向下的熔融吸热峰。未进行任何共混的PLA的玻璃化转变温度为65.1 ℃,其余四组在加入天然橡胶或共聚物后,由于天然橡胶分子链的运动能力比PLA链段要强,后者受到前者的带动,因此导致玻璃化转变温度均下降到了62 ℃。聚乳酸的结晶温度为115.6 ℃,加入天然橡胶后,结晶温度下降明显,这是橡胶颗粒与聚乳酸不相容,颗粒诱导结晶导致的。加入1%嵌段共聚物后,相容情况略好转,结晶温度升高。加入3%共聚物后,结晶温度上升明显,这是因为聚乳酸与天然橡胶相容良好,体系中均相成核更多。从图6中还可以看出,相容剂的引入使共混体系熔融温度降低。最后一种可能是因为加入的10%嵌段共聚物较多,使得可形成PLA晶区的量减少,结晶不完善,且分子链运动能力较强,故而结晶焓和熔融焓也比较低。
以上结果表明,PLA-b-PI和天然橡胶的引入使聚乳酸的热性能发生了变化,基本表现为Tg、Tc和Tm温度的降低,但是降低幅度很小,对其热稳定性影响不大,可放心使用。
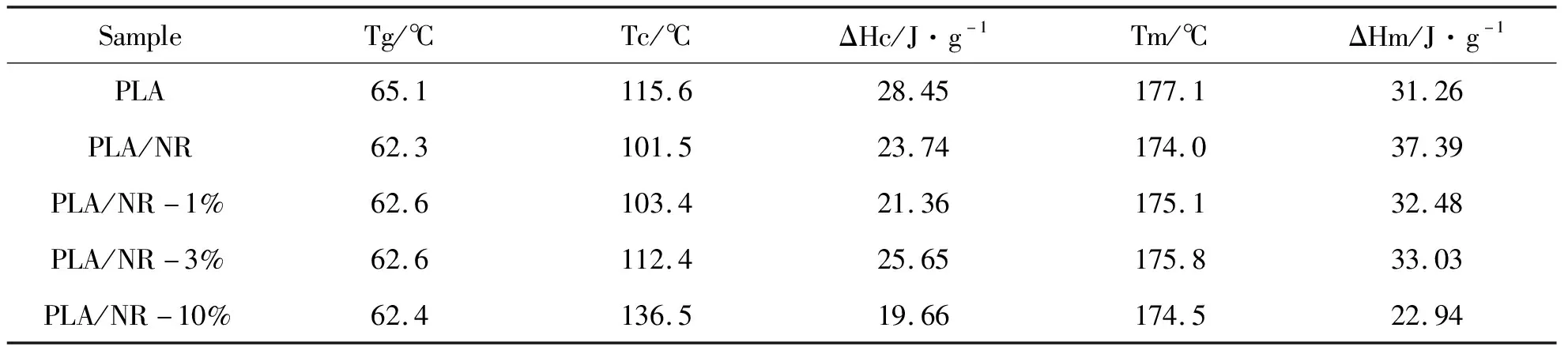
表2 PLA、PLA/NR、PLA/NR-1%、PLA/NR-3%及PLA/NR-10%的DSC数据
注:Tg为玻璃化转变温度;Tc为结晶温度;Tm为熔点;ΔHc为结晶焓;ΔHm为熔融焓
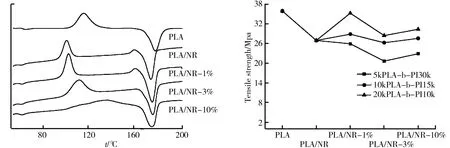
图6 PLA、PLA/NR、PLA/NR-1%、PLA/NR-3%及PLA/NR-10%的DSC曲线,向上放热(exo) 图7 PLA、PLA/NR、PLA/NR-1%、PLA/NR-3%及PLA/NR-10%的拉伸强度
2.5 力学性能表征
选择5kPLA-b-PI10k、10kPLA-b-PI15k和20kPLA-b-PI10k链段的聚合物分别加入到PLA/NR中共混,加入量都是1%、3%和10%,所测拉伸强度、断裂伸长率和冲击强度如图7~图9所示。
5kPLA-b-PI30k、10kPLA-b-PI15k、20kPLA-b-PI10k加入到PLA/NR后的拉伸强度如图7所示,拉伸速率为20 mm/min。由图7可知,纯PLA的拉伸强度是最大的,测得为35.85 MPa;未加入聚合物的PLA/NR为26.9 MPa;加入1%这三种聚合物的拉伸强度和未加入聚合物的PLA/NR差不多,比单纯的PLA要低,分别为25.88 MPa、28.83 MPa和35.20 MPa;加入3%这三种聚合物的拉伸强度相比前一组低一点,分别为20.61 MPa、26.24 MPa和28.40 MPa;加入10%这三种聚合物的拉伸强度分别为22.88 MPa、27.56 MPa和30.31 MPa,都比纯PLA的降低了不少。
PLA、PLA/NR、PLA/NR-1%、PLA/NR-3%及PLA/NR-10%的断裂伸长率如图8所示。由图8可知,纯PLA的最短,仅为5.1%。未加聚合物的PLA/NR断裂伸长率为15.3%;加入1%这三种聚合物的断裂伸长率分别为11.6%、21.4%和22.1%;加入3%这三种聚合物的断裂伸长率分别为9.2%、23.3%和18.0%;加入10%这三种聚合物的断裂伸长率分别为20.7%、14.4%和25.5%。对比可知,加入聚合物后的PLA的断裂伸长率相比纯PLA,达到了原来的3~4倍。
PLA、PLA/NR、PLA/NR-1%、PLA/NR-3%及PLA/NR-10%的冲击强度如图9所示。由图9可知,纯PLA的冲击强度为2.0 kJ/m2;未加聚合物的PLA/NR冲击强度为3.2 kJ/m2;加入1%这三种聚合物的冲击强度分别为3.2 kJ/m2、3.4 kJ/m2和3.7 kJ/m2;加入3%这三种聚合物的冲击强度分别为4.2 kJ/m2、4.4 kJ/m2和4.2 kJ/m2;加入10%这三种聚合物的冲击强度分别为4.2 kJ/m2、3.3 kJ/m2和4.5 kJ/m2。对比可知,只加入天然橡胶,PLA冲击韧性的提升比较有限,加入聚合物后的PLA的冲击强度提升地更加明显,达到了原来的两倍以上。
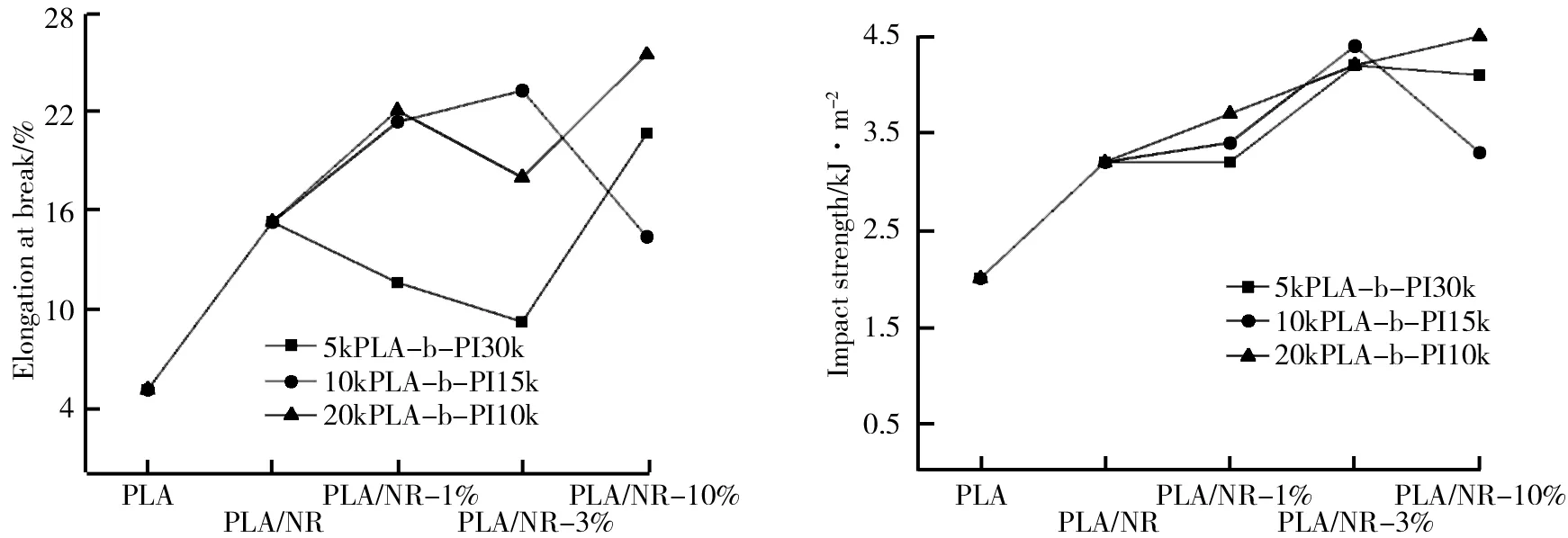
图8 PLA、PLA/NR、PLA/NR-1%、PLA/NR-3%及PLA/NR-10%的断裂伸长率 图9 PLA、PLA/NR、PLA/NR-1%、PLA/NR-3%及PLA/NR-10%的冲击强度
3 结论
研究通过RAFT聚合的方法成功地合成了嵌段聚合物PLA-b-PI,所得聚合物分子量可控,分子量分布指数控制在1.6左右,其结构组成明确且可调整。反应所用的试剂都廉价易得,反应条件也很温和,容易达到。研究还将合成的嵌段共聚物加在了PLA/NR中作相容剂,探究了它对PLA/NR的增容增韧效果,从而确定它的实际应用价值。经过研究发现,PLA-b-PI嵌段共聚物对PLA/NR混合体系的增容效果良好。聚合物加入量在10%时,增容效果最好。在增韧效果上,主要依靠的是天然橡胶,因为天然橡胶是一种弹性体,加入后使PLA的韧性提升到了原来的3~4倍,抗冲能力的提升稍弱一点,这是天然橡胶与聚乳酸不相容造成的。而嵌段共聚物的加入则使PLA/NR的抗冲能力,即冲击韧性的提升更加稳定,保持在了原来的2倍。在热性能上,共聚物和天然橡胶的加入使聚乳酸的Tg、Tc和Tm都下降了一点,所降幅度很小,整体上对聚乳酸的热稳定性影响不大,可以放心使用。积极开发、研制可生物降解的高分子材料,符合我国可持续发展道路的战略国情。PLA是当代最有前景的生物降解塑料之一,因此改性聚乳酸,扩大其应用领域和范围,对于缓解“白色污染”、能源危机等问题,实现全球经济可持续发展具有重要的社会意义和价值。下一步将研究三嵌段聚合物的合成与性质。
