生物信息学方法预测增强子及其作用位点综述
2020-07-04章天骄王亚东
章天骄 王亚东

摘要:对于多细胞真核生物来说,细胞的特异性功能是十分重要的。这就要求在相同遗传物质的基础上,细胞能够通过不同的基因表达模式来适应环境的变化。基因表达调控的因素有很多,近年来随着对基因组非编码区的研究,发现了一些非编码的DNA序列对于基因表达调控具有重要意义。增强子是对基因表达调控具有重要作用的非编码序列元件之一。一些增强子能够通过转录产生具有调控功能的RNA,也被称为增强子RNA(enhancer RNA,eRNA)。因此对于增强子的序列特征、作用位点以及在特定时间和特定组织中表达模式的研究成为了基因表达调控领域的一个重要问题。
关键词: 增强子; eRNA; 基因表达调控
【Abstract】 Specific function of cells is very important for multicellular eukaryotes. Despite the same genetic material, cells can adapt to environment changes through different gene expression patterns. There are many factors in gene expression regulation. In recent years, with the study of genomic non-coding region, it has been found that some non-coding DNA sequences are important for gene expression regulation. Enhancers are one of the non-coding sequence components that play an important role in gene expression regulation. Some enhancers can be transcribed into RNA with regulatory function, also known as enhancer RNA (eRNA). Therefore, the study on enhancer sequence features, target genes, and expression patterns in specific time and specific tissues has become an important issue in the field of gene expression regulation.
【Key words】 enhancer; eRNA; gene expression regulation
0引言
增強子是DNA序列上增强基因表达的顺式调控元件,通常位于转录起始位点较远的位置。与启动子不同,增强子对基因表达的调控作用具有高度明显的组织特异性。增强子对基因表达的调控不是“开关”模式,而是一种可变调控,即影响基因表达量的高低,而不是直接关闭或开启表达的调控方式[1]。
增强子最初于1981年由Banerji和Moreau等人在猿猴空泡病毒40(SV40)的基因组中被发现[2]。1983年在小鼠免疫重链基因中发现了第一个非病毒的增强子[3]。哺乳动物中增强子的数目为50 000到100 000个。大部分增强子位于内含子区和基因间区,少部分位于外显子区[4]。在相近的基因组区域内多个增强子聚集成簇的现象被称为超级增强子或增强子簇。研究发现其横跨很大的基因组区同时富集了大量的转录因子及转录中介复合物[5]。超级增强子经常位于细胞特异性功能基因的附近,并且富含细胞特异性转录因子结合序列模体。虽然超级增强子被广泛应用于多个研究中,但是却没有一个清晰的定义[6]。
[BT4]1增强子概述
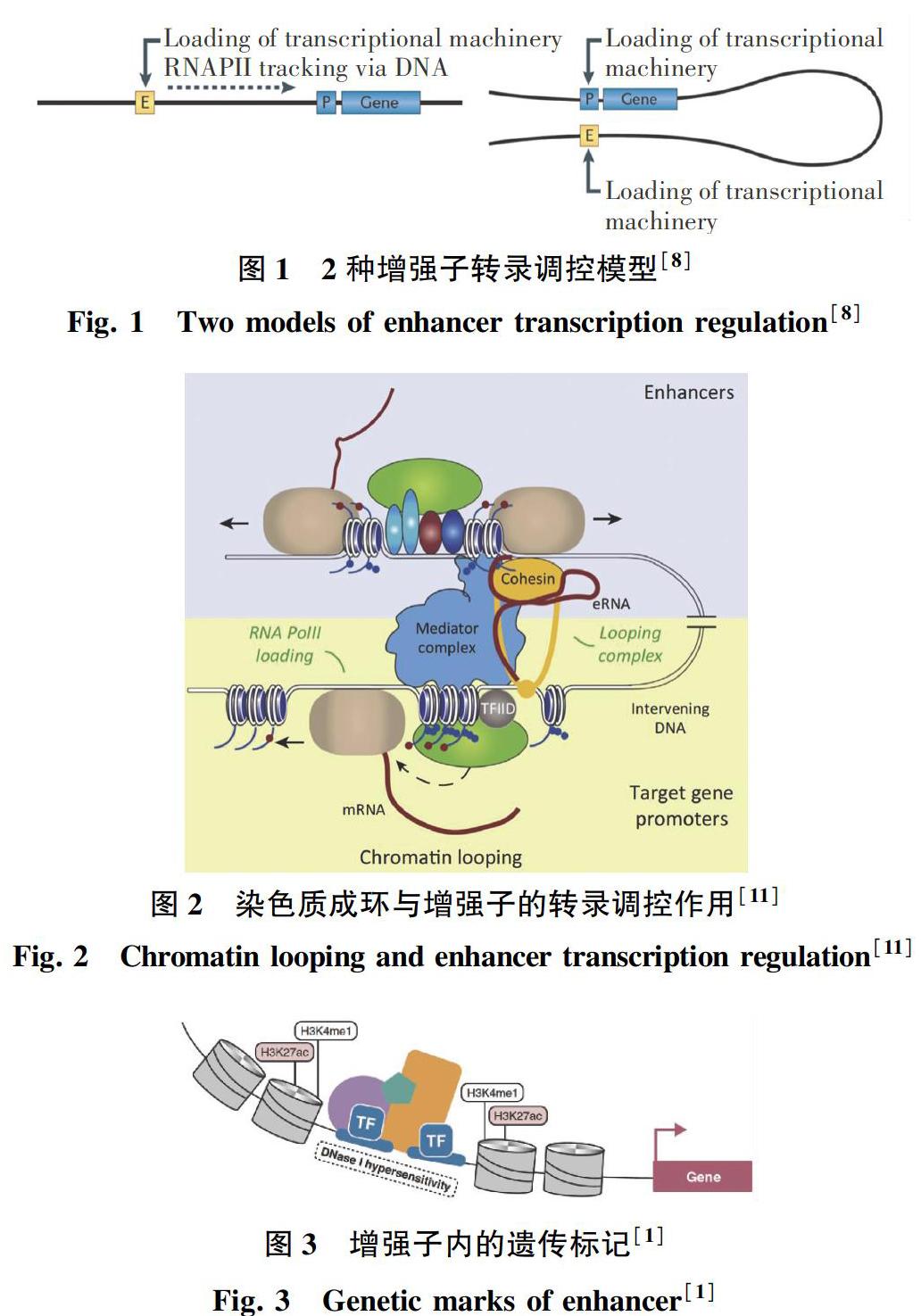
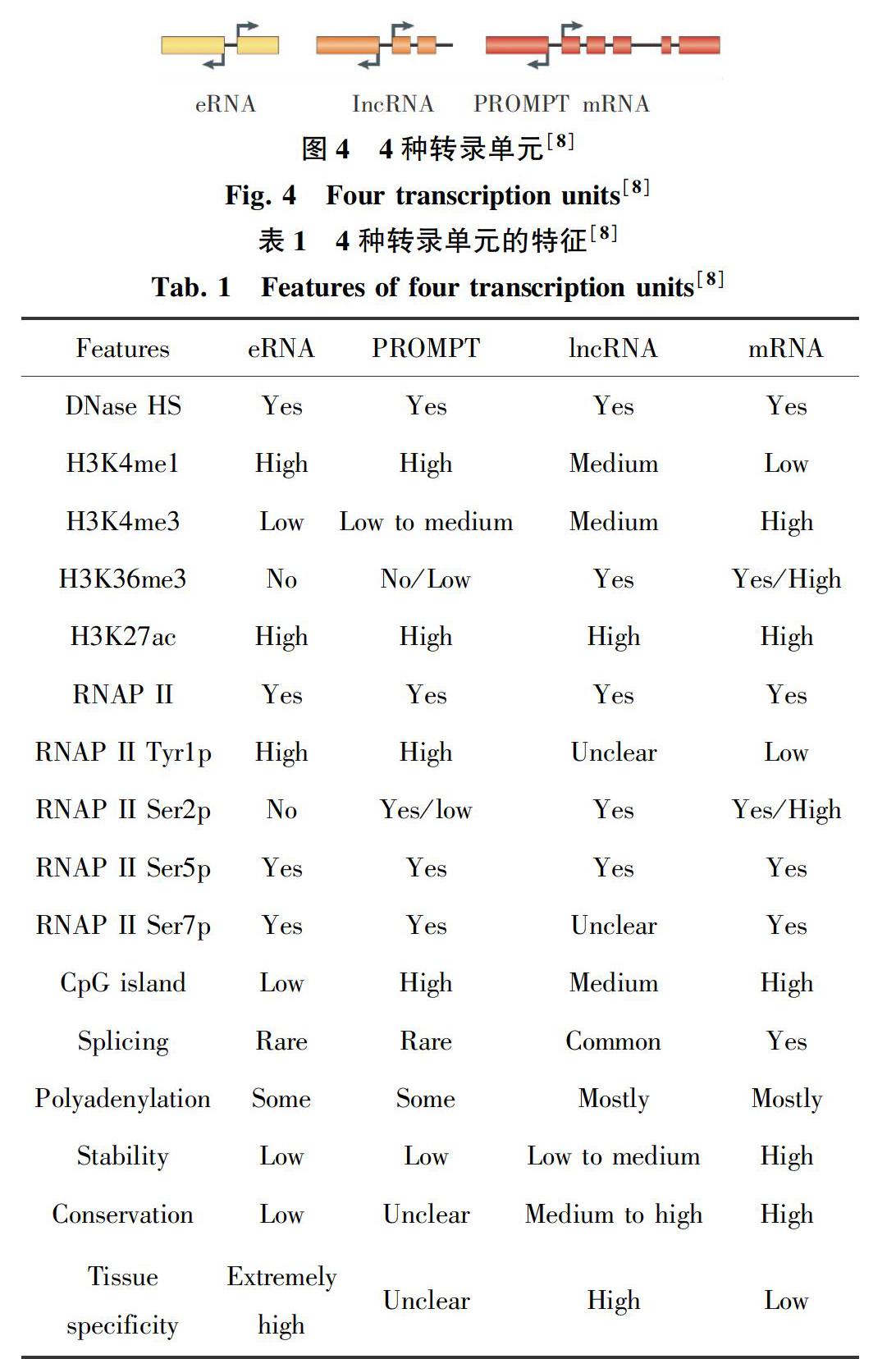
增强子通过与其目标基因启动子相互作用实现对基因的表达调控。这种相互作用,可能是顺式(in cis)作用,也可能是反式(in trans)作用。顺式作用是指增强子及其作用位点基因在同一条染色体上,反式作用则指增强子及其作用位点基因在不同的染色体上[7]。目前对增强子调控基因表达有2种模型,如图1所示[8]。第一种是轨道调控模型,即RNA聚合酶II及其转录复合物沿着DNA轨道从增强子到启动子滑动。虽然这种模型在一些例子中被证实是存在的[9],但过去二三十年的研究更加支持另一种模型。第二种模型是环状调控模型,增强子通过染色质成环现象与其调控基因的启动子区域相互临近[10]。图2显示了增强子通过染色质成环与启动子临近调控基因表达的现象[11]。
增强子内部包含多种遗传标记位点,最常见的是转录因子结合位点。转录因子与转录复合物的辅助激活因子p300/CBP通过相互作用富集在增强子区域内。这些与转录因子结合的区域内核小体的结合度显著下降,导致容易被脱氧核糖核酸酶I(Deoxyribonuclease I,简称DNase I)剪切。这些核小体缺失区域(Nucleosome-depleted Regions,NDRs)两侧被特殊的组蛋白修饰所标记,例如H3K4me1和H3K27ac。H3K4me1与不活跃和活跃的增强子均相关联,H3K27ac只与活跃的增强子相关联。图3显示了增强子内部常见的遗传标记[1]。
第一次发现增强子的转录现象是在β-珠蛋白的基因座控制区(Locus Control Region,LCR),随着高通量测序技术的广泛应用,增强子的转录被发现是一种普遍现象。2010年,Kim等人[12]对小鼠的中枢神经细胞中剔除rRNA后的其它RNA测序时,发现了增强子RNA的双向转录现象。这种由增强子经过转录过程表达出的RNA为eRNA。这个发现使得人们认识到增强子区域不仅富集转录因子,更是一个转录激活区。与之前的研究一致,增强子区域同样富集转录起始复合物,例如RNA聚合酶II[13]。
eRNA的长度通常较长,因此eRNA经常被认为是长非编码RNA(Long Noncoding RNA,lncRNA)的子集。然而,一部分eRNA却不在lncRNA的数据库中[14]。造成这种现象有2层原因,其一是eRNA通常是通过其转录区域具有增强子遗传标记所发现的[14],而lncRNA则是根据其长度大于200 bp所定义的[14];其二是eRNA由于其不稳定性或者表达量较低没能被构建lncRNA数据库的方法所识别[14]。由于这种现象导致eRNA和lncRNA存在交集,故而将交集部分定义为lnc-eRNA。
eRNA作为一种新的转录单元,与其它的转录单元既有相似性也有不同之处。图4列出了4种常见的转录单元[8]。其中,启动子上游转录区(Promoter Upstream Transcripts,PROMPTs)由于与eRNA具有相似的性质而被单独列出来。这四种常见转录单元的部分性质及特征对比见表1[8]。
2增强子预测
对于增强子预测的研究是所有相关增强子研究的基础问题。只有在基因组上识别出增强子所在的位置后,才能对增强子的其它性质及功能进行研究。大量的研究使用了不同的生物数据来预测增强子的位置。这些生物数据主要分为5种类别,详述如下。
(1)使用了序列保守性数据和转录因子结合位点数据进行计算学分析。Woolfe等人[15]、Pennacchio等人[16]和Visel等人[17]对不同物种间非编码元件进行保守性分析来预测增强子。其中,Pennacchio等人[16]对人类和红鳍东方鲀的基因组进行了序列比对,找到保守的非编码区域。然后对这些区域进行模式序列的搜索,找到2个物种共有的具有增强基因表达程度的模式序列。然后,对于所有未知功能的保守非编码区域进行打分,对应数学公式可写为:
Wasserman等人[18]将转录因子结合位点和保守性分析相结合,对不同物种的非编码区域进行分析来预测增强子。这对于已知结合序列模式信息的转录因子能够很好地预测其结合的基因组位置,但同时也包含了其它非增强子的与转录因子结合的调控元件序列,因此结果具有很高的假阳性。
(2)使用了调控因子的结合数据,包括转录因子的ChIP-seq数据和转录辅激活物p300的ChIP-seq数据。Chen等人[19]和Zinzen等人[20]使用了转录因子的ChIP-seq数据来预测增强子。Chen等人[19]使用了13种转录因子(Nanog,Oct4,STAT3,Smad1,Sox2,Zfx,c-Myc,n-Myc,Klf4,Esrrb,Tcfcp2l1,E2f1和CTCF)和2种转录调控元件(p300和Suz12)对胚胎干(Embryonic Stem,ES)细胞的转录调控网络进行构建。这种方法能够识别那些与已知转录因子结合的增强子。但是这种方法需要知道具体的转录因子来进行实验设计。同时也不能区分增强子和启动子区域,因为这些区域都会结合转录因子。另一方面也不是所有的增强子都与转录因子相结合。
Visel等人[21]和May等人[22]使用了转录辅激活物p300的ChIP-seq数据来预测增强子。Visel等人[21]对全基因组p300的ChIP-seq数据进行分析得到了p300的富集位置。然后针对这些p300富集的位置是否具有增强子活性进行检测,发现约88%的位置具有增强子的活性。并且发现这些p300富集的位置大多是保守的。这种方法被广泛地应用于增强子位置的预测,但对于活跃的增强子和不活跃的增强子的区分效果不好。
(3)是与染色质的可及性(Chromatin Accessibility)相关的数据。染色质的可及性是指染色质缠绕的核小体从致密变为松散,导致转录调控元件可以顺利结合到其上面起调控作用的性质。应用染色质的可及性来识别增强子主要有以下3种技术。Dorschner等人[23]使用了脱氧核糖核酸酶I超敏感位点测序(DNase I Hypersensitive Sites Sequencing,DNase-seq)数据。Giresi等人[24]使用了甲醛辅助分离调控元件测序(Formaldehyde-assisted Isolation of Regulatory Elements Sequencing,FAIRE-seq)数据。Buenrostro等人[25]使用了转座酶可及染色质测序(Assay for Transposase-accessible Chromatin Sequencing,ATAC-seq)数据。DNase-seq需要使用大量的细胞用于实验,而ATAC-seq需求的细胞量则很少,同时实验周期也相对较短。但是应用染色质可及性数据进行预测同样也会使结果存在假阳性,即会有其他转录调控单位,例如启动子、隔离子和沉默子等被包含进结果中。
(4)是组蛋白修饰数据。Heintzman等人[26]应用了H3K4me1和H3K27ac来预测增强子的位置,这两种组蛋白修饰前者是与增强子特定相关,后者则是与激活的调控区相关,同时应用这两种组蛋白修饰预测得到的基因组调控区域便是激活的增强子区域。此外还有多种组蛋白修饰与DNA序列调控元件的关联关系:H3K4me3与启动子相关联,H3K4me2与启动子和增强子都相关,H3K9ac也与激活的调控区相关,H3K36me3和H4K20me1与转录区相关,H3K27me3与多梳抑制区域相关等。这种预测方法的优点是不同物种间组蛋白修饰数据来源广泛,能够有效地辅助不同需求的研究,缺点是全基因组范围内组蛋白修饰信号十分广泛,不利于高精度预测增强子位置。
(5)是基于eRNA数据进行预测。上文中已经提到,增强子会转录出eRNA,这部分eRNA通过测序技术被检测到再映射回原基因组就能得到增强子的位置信息。Kim等人[12]使用了RNA-seq技术,这种技术的优点是eRNA和其附近的基因表达水平可以同时被量化,缺点是低表达水平的eRNA不会被检测到。Lai等人[27],Melgar等人[28],Mayer等人[29]分别使用了染色质关联RNA测序(Chromatin-associated RNA-seq,ChAR-seq)、GRO-seq和NET-seq技术来检测eRNA,这三种技术的优势是都可以检测不稳定的eRNA。Andersson等人[30]使用了CAGE技术来检测eRNA。这种技术的优点在于可以高精度地确定eRNA的转录起始位点,缺点是对于检测表达量较低的eRNA需要的样本量较大。同时所有基于eRNA數据确定增强子位置的方法都不能用于预测未表达的增强子。
综上,不同的增强子预测方法在本质上是使用了增强子附近不同的生物信号数据。图5比较了不同增强子识别方法的差异[31]。
3增强子作用位点预测
增强子的作用位点指的是增强子对基因表达起增强作用。这种作用体现在增强子与基因的启动子区域相互作用,从而调控基因的表达。通常认为增强子与启动子的相互作用是通过物理上的成环结构实现的。这种物理上的近邻会募集多种转录因子和转录辅助因子结合到增强子和启动子区域。而这些转录因子等则会吸引RNA聚合酶从而引起转录的发生。因此要研究一个增强子的生物学功能,确定其作用位点是至关重要的。目前对于增强子作用位点的研究主要分为2类方法:基于生物学实验的方法和基于计算学的方法。随着生物实验技术的进步,从生物学实验的角度来研究增强子的作用位点变得准确可靠,但实验成本也随之增高。基于计算学的方法虽然不如实验方法准确,但其高通量的特性和较低的实验成本能够很好地辅助生物学实验的进行。
由于基于生物实验的方法预测增强子作用位点的成本较高,因此需要计算学方法的辅助。近年来由于生物信息學的发展,一系列基于不同增强子特征的计算学预测方法被开发出来。这些计算学方法都需要比较不同细胞类型下增强子附近调控信号的分布模式来预测增强子与基因的关系。
最早被应用于预测增强子与基因调控关系的特征就是基因与增强子间的基因组距离。在定位一个增强子位置后,在基因组上距离其最近的基因被认为是该增强子的调控基因。由于增强子本身具有超远距离调控的性质,这种预测方法的准确率通常不高且变化幅度很大,错误发现率(False Discovery Rate,FDR)约为40%~73%。由于增强子是对基因表达起增强调控作用,为了提高预测的准确性,研究人员将增强子的调控基因定位为距离其最近的表达基因。这种方案需要用到基因表达数据,其准确性仍然较低,FDR值约为53%~77%。
Ernst等人[32]考察了人类9种细胞系中增强子附近的组蛋白修饰数据H3K4me1,H3K4me2和H3K27ac。通过对125 kb距离内基因的RNA-seq表达数据进行关联分析,寻找具有共同变化模式的“增强子-基因”对,来预测增强子的作用位点。这种预测方法使用了增强子的组蛋白修饰数据作为特征与基因表达数据关联,能够一定程度提高预测的准确度。但由于距离的限制只能预测增强子附近125 kb范围内的作用位点。
这种预测方法使用了RNA pol II作为基因启动子区域的标志,使用H3K4me1作为增强子区域的标志,同时使用H3K27ac作为转录活跃区的标志,通过关联分析,得到增强子与启动子共有的组蛋白修饰变化模式,以此说明二者存在关联关系。使用组蛋白修饰数据能够避免上述依赖表达量方法只能预测处于活跃状态基因的问题。但是由于组蛋白修饰数据本身在基因组中十分广泛,因此预测精度也会受到一定限制。
Andersson等人[30]使用了CAGE数据来预测增强子的作用位点。通过CAGE可以测得增强子和基因TSS的表达量,选取距离在500 kb范围内表达量在1 TPM以上的增强子和启动子,计算二者的皮尔森相关系数并进行假设检验。由于上文已经阐述了增强子能够通过转录产生eRNA对目标基因起调控作用,因此可以用二者表达量的相关性来预测关联关系。这种基于表达量的方法预测增强子作用位点的优点是能够反映活跃增强子与其调控基因的相关性,但同时有很多增强子的表达量较低、甚至没有表达,这些增强子的作用位点就难以用基于表达量的方法预测,同时500 kb的距离也限制了预测的准确度。
Corradin等人[36]和Factor等人[37]分别开发了PreSTIGE和PreSTIGEouse来预测人类和小鼠中增强子的作用位点。这两种方法都使用了组蛋白修饰数据H3K4me1来标识增强子的位置。通过分析与附近基因的表达量相关性确定关联关系。与以往固定距离方法不同,Corradin等人[36]使用了转录因子CTCF的位置数据作为确定增强子与基因间距离的参考。CTCF是与隔离子活性相关的转录因子,在基因组中起分割作用。增强子只能在隔离子分割的区域内对基因的表达起增强作用。与将最近基因作为增强子作用位点的方法相比,使用CTCF数据可以有效降低FDR,其值为13%~23%。这种方法能够一定程度避免由于距离的不确定性导致预测性能较低的现象。缺点是由于只使用了一种组蛋白修饰数据作为增强子的标识,导致预测结果的准确性仍有一定的不足。
He等人[38]开发了IM-PET,应用多种遗传特征的组合来预测增强子的作用位点。研究中根据增强子附近不同遗传特征的类型将所有特征分为4个类别,对此可做阐释分述如下。
(1)是反映增强子与启动子表达活跃度的相关性特征。首先通过组蛋白修饰数据H3K4me1、H3K27ac和H3K4me3估计增强子的表达活性,然后与启动子的RNA-seq数据进行关联分析,计算皮尔森相关系数作为第一类特征,简称EPC。
(2)是反映转录因子与目标启动子关联关系的特征。由于第一类特征只能反映在DNA序列层次上的调控,没有反映在转录因子层次上的调控关系,因此需要构建转录因子在增强子区域的结合度与基因表达的关联关系。通过计算二者的皮尔森相关系数作为第二类特征,简称TPC。
(3)是反映启动子和增强子的保守性的特征。由于启动子和增强子作为调控区域的保守性在序列层次上可能不强,但在同线性的层次上却有着较高的保守性,因此可以分别计算一定距离内启动子和增强子区域在多物种中的保守性得分,将得分标准化后的乘积作为第三类特征,简称COEV。
(4)是反映启动子和增强子间距离远近的特征。即转录起始位点到增强子中心的距离作为第四类特征,简称DIS。
在完成特征集合的构建后,根据已有的训练样本集训练随机森林分类器。然后将此分类器应用到测试样本集上检测性能调整分类器参数。最后对给定的增强子-启动子对数据,应用分类器来预测二者的关联性。图6显示了应用IM-PET预测增强子与启动子关联关系的流程图。
IM-PET综合使用了多种特征来分析和预测增强子与启动子间的关联关系。比起前人单纯使用一种或几种特征能够从生物学角度更加全面地描述强子与启动子间的关系。同时由于使用了机器学习的方法对分类器进行训练,使得FDR大大降低(约1%),并且可分析的基因组距离大大增加(2 Mb),有效提高了预测精度。但是由于预测方法中对于特征集合的构建使用了较多的特征,并且缺少对这些特征重要程度的描述,使得一些与预测关联性不强的特征也纳入进来,预测结果也会受到一定影响。
以上从计算学的角度总结了预测增强子作用位点的方法。这些方法大多围绕着提取增强子与启动子附近的相关特征,构建特征值间的关联关系来预测增强子的作用位点。基于计算学的方法能够高效经济地识别潜在的增强子与基因的相互作用关系,但是对于增强子与启动子的反式作用则显得乏力。这些应用特定细胞条件下特征一致性变化的预测方法难以实现对所有类型细胞中均表达的管家基因的预测。因此,这些计算学的方法都给出了生物实验验证增强子与启动子的关联关系。
4结束语
增強子是基因表达调控的重要元件之一。对于增强子本身的识别及其作用位点的预测一直是相关领域的研究热点问题。近年来生物数据监测技术的不断进步带来了海量的生物数据,同时生物信息技术的发展为研究增强子的生物学功能提供了强大的技术手段。本文总结了目前生物信息领域对增强子相关问题的研究热点,着重总结了增强子及其作用位点预测的研究方法。
参考文献
[1] CORRADIN O, SCACHERI P C. Enhancer variants: Evaluating functions in common disease[J]. Genome Medicine, 2014, 6(10): 85.
[2] BANERJI J, RUSCONI S, SCHAFFNER W. Expression of a beta-globin gene is enhanced by remote SV40 DNA sequences[J]. Cell, 1981, 27(2 Pt 1): 299.
[3] BANERJI J, OLSON L, SCHAFFNER W. A lymphocyte-specific cellular enhancer is located downstream of the joining region in immunoglobulin heavy chain genes[J]. Cell, 1983, 33(3): 729.
[4] BIRNBAUM R Y, CLOWNEY E J, AGAMY O, et al. Coding exons function as tissue-specific enhancers of nearby genes[J]. Genome Research, 2012, 22(6): 1059.
[5] HNISZ D, ABRAHAM B J, LEE T I, et al. Super-enhancers in the control of cell identity and disease[J]. Cell, 2013, 155(4): 934.
[6] POTT S, LIEB J D. What are super-enhancers?[J]. Nature Genetics, 2015, 47(1): 8.
[7] SASAKI-IWAOKA H, MARUYAMA K, ENDOH H, et al. A trans-acting enhancer modulates estrogen-mediated transcription of reporter genes in osteoblasts[J]. Journal of Bone and Mineral Research: The Official Journal of the American Society for Bone and Mineral Research, 1999, 14(2): 248.
[8] LI W, NOTANI D, ROSENFELD M G. Enhancers as non-coding RNA transcription units: Recent insights and future perspectives[J]. Nature Reviews. Genetics, 2016, 17(4): 207.
[9] HATZIS P, TALIANIDIS I. Dynamics of enhancer-promoter communication during differentiation-induced gene activation[J]. Molecular Cell, 2002, 10(6): 1467.
[10]WANG Qianben, CARROLL J S, BROWN M. Spatial and temporal recruitment of androgen receptor and its coactivators involves chromosomal looping and polymerase tracking[J]. Molecular Cell, 2005, 19(5): 631.
[11]LAM M T Y, LI Wenbo, ROSENFELD M G, et al. Enhancer RNAs and regulated transcriptional programs[J]. Trends in Biochemical Sciences, 2014, 39(4): 170.
[12]KIM T K, HEMBERG M, GRAY J M, et al. Widespread transcription at neuronal activity-regulated enhancers[J]. Nature, 2010, 465(7295): 182.
[13]KOCH F, FENOUIL R, GUT M, et al. Transcription initiation platforms and GTF recruitment at tissue-specific enhancers and promoters[J]. Nature Structural & Molecular Biology, 2011, 18(8): 956.
[14]DERRIEN T, JOHNSON R, BUSSOTTI G, et al. The GENCODE v7 catalog of human long noncoding RNAs: Analysis of their gene structure, evolution, and expression[J]. Genome Research, 2012, 22(9): 1775.
[15]WOOLFE A, GOODSON M, GOODE D K, et al. Highly conserved non-coding sequences are associated with vertebrate development[J]. PLoS biology, 2005, 3(1): 116.
[16]PENNACCHIO L A, AHITUV N, MOSES A M, et al. In vivo enhancer analysis of human conserved non-coding sequences[J]. Nature, 2006, 444(7118): 499.
[17]VISEL A, PRABHAKAR S, AKIYAMA J A, et al. Ultraconservation identifies a small subset of extremely constrained developmental enhancers[J]. Nature Genetics, 2008, 40(2): 158.
[18]WASSERMAN W W, FICKETT J W. Identification of regulatory regions which confer muscle-specific gene expression[J]. Journal of Molecular Biology, 1998, 278(1): 167.
[19]CHEN Xi, XU Han, YUAN Ping, et al. Integration of external signaling pathways with the core transcriptional network in embryonic stem cells[J]. Cell, 2008, 133(6): 1106.
[20]ZINZEN R P, GIRARDOT C, GAGNEUR J, et al. Combinatorial binding predicts spatio-temporal cis-regulatory activity[J]. Nature, 2009, 462(7269): 65.
[21]VISEL A, BLOW M J, LI Zirong, et al. ChIP-seq accurately predicts tissue-specific activity of enhancers[J]. Nature, 2009, 457(7231): 854.
[22]MAY D, BLOW M J, KAPLAN T, et al. Large-scale discovery of enhancers from human heart tissue[J]. Nature Genetics, 2011, 44(1): 89.
[23]DORSCHNER M O, HAWRYLYCZ M, HUMBERT R, et al. High-throughput localization of functional elements by quantitative chromatin profiling[J]. Nature Methods, 2004, 1(3): 219.
[24]GIRESI P G, KIM J, MCDANIELL R M, et al. FAIRE (Formaldehyde-Assisted Isolation of Regulatory Elements) isolates active regulatory elements from human chromatin[J]. Genome Research, 2007, 17(6): 877.
[25]BUENROSTRO J D, GIRESI P G, ZABA L C, et al. Transposition of native chromatin for fast and sensitive epigenomic profiling of open chromatin, DNA-binding proteins and nucleosome position[J]. Nature Methods, 2013, 10(12): 1213.
[26]HEINTZMAN N D, STUART R K, HON G, et al. Distinct and predictive chromatin signatures of transcriptional promoters and enhancers in the human genome[J]. Nature Genetics, 2007, 39(3): 311.
[27]LAI F, GARDINI A, ZHANG A, et al. Integrator mediates the biogenesis of enhancer RNAs[J]. Nature, 2015, 525(7569): 399.
[28]MELGAR M F, COLLINS F S, SETHUPATHY P. Discovery of active enhancers through bidirectional expression of short transcripts[J]. Genome Biology, 2011, 12(11): R113.
[29]MAYER A, DI IULIO J, MALERI S, et al. Native elongating transcript sequencing reveals human transcriptional activity at nucleotide resolution[J]. Cell, 2015, 161(3): 541.
[30]ANDERSSON R, GEBHARD C, MIGUEL-ESCALADA I, et al. An atlas of active enhancers across human cell types and tissues[J]. Nature, 2014, 507(7493): 455.
[31]MURAKAWA Y, YOSHIHARA M, KAWAJI H, et al. Enhanced identification of transcriptional enhancers provides mechanistic insights into diseases[J]. Trends in genetics: TIG, 2016, 32(2): 76.
[32]ERNST J, KHERADPOUR P, MIKKELSEN T S, et al. Mapping and analysis of chromatin state dynamics in nine human cell types[J]. Nature, 2011, 473(7345): 43.
[33]THURMAN R E, RYNES E, HUMBERT R, et al. The accessible chromatin landscape of the human genome[J]. Nature, 2012, 489(7414): 75.
[34]SHEFFIELD N C, THURMAN R E, SONG Lingyun, et al. Patterns of regulatory activity across diverse human cell types predict tissue identity, transcription factor binding, and long-range interactions[J]. Genome Research, 2013, 23(5): 777.
[35]SHEN Yin, YUE Feng, MCCLEARY D F, et al. A map of the cis-regulatory sequences in the mouse genome[J]. Nature, 2012, 488(7409): 116.
[36]CORRADIN O, SAIAKHOVA A, AKHTAR-ZAIDI B, et al. Combinatorial effects of multiple enhancer variants in linkage disequilibrium dictate levels of gene expression to confer susceptibility to common traits[J]. Genome Research, 2014, 24(1): 5.
[37]FACTOR D C, CORRADIN O, ZENTNER G E, et al. Epigenomic comparison reveals activation of “seed” enhancers during transition from naive to primed pluripotency[J]. Cell Stem Cell, 2014, 14(6): 854.
[38]HE Bing, CHEN Changya, TENG Li, et al. Global view of enhancer-promoter interactome in human cells[J]. Proceedings of the National Academy of Sciences of the United States of America, 2014, 111(21): E2191.
