5-氮-2′-脱氧胞苷对人肠癌细胞侵袭性研究及机制探讨
2020-07-01方晓明邵银灿郭胜才娄敏峰王喜姚宁
方晓明 邵银灿 郭胜才 娄敏峰 王喜 姚宁
解放军联勤保障部队第903 医院1普通外科,2肿瘤科(杭州310004)
结直肠癌在我国是常见的恶性肿瘤之一,其发病率已居第三并仍呈上升趋势。众多研究结果表明:人肠癌细胞的发生发展及无限增殖与多个癌基因的激活和抑癌基因的失活有关[1-2],其中包含有细胞周期调控因子—细胞周期素依赖性激酶抑制因子(cyclin-dependent kinases,CKIs)的表达沉默,而这种抑癌基因的失活和沉默与肿瘤细胞内DNA 甲基转移酶(DNA methyltransferase,DNMTs)过表达所致基因启动子区过度甲基化相关[3]。笔者在前期的研究结果已发现:人肠癌RKO 细胞CKIs 的激酶4 抑制因子家族(inhibitors of kinase 4、INK4、p15ink4b及p16ink4a/CDKN2)基因启动子区5′CpG 岛呈高甲基化状态,且这种异常的高甲基化具有可逆性[4-5],但对其逆转后的增殖侵袭能力尚未明确。故本研究旨在通过应用特异性DNMTs 抑制剂5-氮-2′-脱氧胞苷(5-Aza-2′-deoxycytidine,5-Aza-CdR)干预人肠癌RKO 细胞,探讨5-Aza-CdR 对INK4 家族基因表达及对人肠癌细胞增殖、迁移和侵袭能力的影响,探索大肠癌治疗的新靶点。
1 材料与方法
1.1 材料人结肠癌细胞株RKO 由浙江大学肿瘤研究所提供。5-Aza-CdR、软琼脂及吖啶橙购自美国Sigma 公司;单克隆抗体p15ink4b及p16ink4a/CDKN2 购自美国Santa Cruz 公司;胎牛血清为杭州江滨生物技术有限公司;DMEM 培养液购自美国GIBCO 公司。
1.2 方法
1.2.1 细胞培养及药物处理人结肠癌细胞株RKO在含10%胎牛血清,100 U/mL青酶素,100 U/mL链酶素的DMEM 高糖(4 500 mg/L)培养基中,置于37 ℃、含体积分数为5%CO2的饱和湿度箱中培养,每4 ~5 d 消化传代1 次。
1.2.2 细胞分组取对数生长期细胞传代24 h 贴壁后,分为经5-Aza-CdR 作用的实验组及未经5-Aza-CdR作用的阴性对照组,实验组分别用1×10-7、5 × 10-7、1 × 10-6mol/L 3 个不同浓度5-Aza-CdR 作用72 h,继续培养,用于后续实验。
1.2.3 MTT 法检测取经上述处理前后的对数生长期细胞,按每孔2 × 103个(200 μL)接种于96孔培养板中,每3 孔为1 组,共7 板,每日1 板加入5 g/L MTT 液20 μL,37 ℃再孵育4 h 后,弃上清,加DMSO 150 μL 振荡溶解,在570 nm 波长酶联免疫检测仪(Opsys MD)上测定各孔吸光度值(A),求其均值,以A值为纵坐标,时间(d)为横坐标绘制生长曲线。细胞增殖抑制率(CPIR)按公式:CPIR=(1-实验组A均值/对照组A均值)×100%计算。
1.2.4 细胞迁移和侵袭实验利用Matrigel 胶人工基底膜和Transwell 小室检测4 组细胞的迁移能力,每组各设3 个复孔,细胞数为5 × 103个/孔。DMEM-HG 培养48 h 后,4%多聚甲醛固定,0.1%结晶紫染色,取微孔膜,倒置显微镜下观察,随机取5 个典型视野观察计数穿膜细胞数,求各视野的平均细胞数表示肿瘤细胞的侵袭迁移能力。
1.2.5 软琼脂克隆细胞集落实验取5%琼脂沸水浴中溶化,将1 份琼脂与9 份预温37℃的DMEMHG 混匀,浇入6 孔培养板中,置室温下凝固制备底层琼脂;胰蛋白酶消化RKO 细胞,分散成单个细胞悬液并计数,调整各组细胞密度至1×103个/mL,置室温备用。将RKO 细胞悬液加入铺有底层琼脂的6 孔培养板中,每组设3 个复孔,每孔1.5 mL,置室温下使琼脂凝固,制备顶层琼脂。CO2孵箱内培养2 周,加入浓度为10 mg/mL 的吖啶橙1 h 后,在倒置显微镜上观察集落形成情况并随机计数5 个典型视野,计算其平均数及标准差。
1.2.6 免疫组织化学染色收集各5-Aza-CdR 浓度处理的结肠细胞,涂片,乙醇固定,一抗p15ink4b和p16/CDKN2 工作浓度为1∶100(Santa Cruz 公司产品),按试剂盒说明常规SP 染色,DAB 显色,光镜观察,以细胞质出现棕黄色颗粒为阳性,比较细胞形态、胞质染色强弱及阳性细胞数。
1.3 统计学方法采用SPSS 19.0统计软件对实验数据进行统计分析。定量数据以描述,先作正态性检验和方差齐性检验,符合正态分布且方差齐,则多组间数据采用单因素方差分析,组内两两比较采用t检验。P<0.05为差异有统计学意义。
2 结果
2.1 细胞生长曲线和细胞增殖抑制率经不同浓度5-Aza-CdR 处理后,各组细胞生长曲线如图1 所示,经统计符合正态分布。与未经5-Aza-CdR 处理的阴性对照组比,3 实验组RKO 细胞的生长增殖活性均有明显抑制,在2、3、4 d 时抑制差异无统计学意义(F=0.642、3.75、8.86,P>0.05);7 d 时阴性对照组、0.1 μmol/L 组、0.5 μmol/L 组、1.0 μmol/L 组细胞的细胞抑制率分别为(0.00 ± 0.99)%、(40.70±0.54)%、(52.00±1.62)%和(62.62±1.40)%,4组比较,差异有统计学意义(F= 347.38,P<0.01)。实验组细胞时间抑制率见图2。7 d 时对照组分别与0.1 μmol/L 组、0.5 μmol/L 组和1.0 μmol/L组比较(t=62.538、47.457、62.398,P<0.01),差异均有统计学意义(图2)。

图1 5-Aza-CdR 对肠癌RKO 细胞生长曲线图Fig.1 Growth curve of 5-Aza-CdR on RKO cell lines
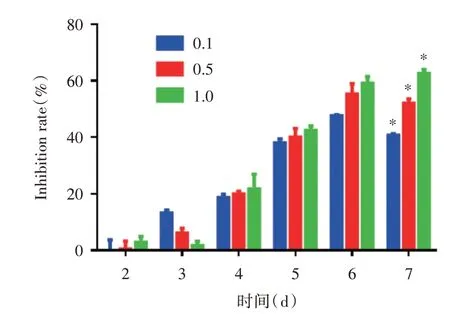
图2 5-Aza-CdR 对肠癌RKO 细胞增殖抑制率的影响Fig.2 Inhibition rate of 5-Aza-CdR on RKO cell lines
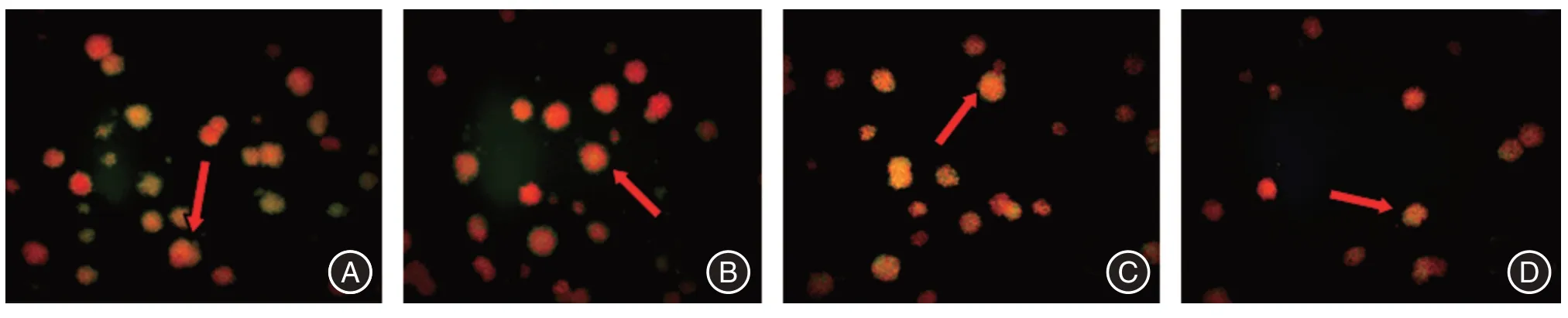
图3 软琼脂克隆(体外)致瘤性试验(吖啶橙染色×200)Fig.3 Soft agar cloning experiment in vitro(acridine orange staining× 200)
2.2 细胞迁移和侵袭能力细胞迁移和侵袭结果显示,7 d 时阴性对照组、0.1 μmol/L 组、0.5 μmol/L组和1.0 μmol/L 实验组细胞迁移数目分别为(67.4±7.2)、(35.3±4.6)、(29.5±7.3)和(25.3±6.2)个。4组比较,差异有统计学意义(F=72.727,P<0.01)。对照组分别与0.1 μmol/L 组(t= 9.344,P<0.05)、0.5 μmol/L 组(t=10.231,P<0.01)和1.0 μmol/L 组(t= 11.589,P<0.01)两两比较显示,差异均有统计学意义。
2.3 克隆细胞集落致瘤性实验结果体外克隆细胞集落实验结果显示,对照组、0.1 μmol/L 组、0.5 μmol/L 组及1.0 μmol/L 组细胞集落数目分别为:(36.8 ± 5.1)、(32.4 ± 7.2)、(21.3 ± 5.4)、(19.5±6.4)个。4组比较,差异有统计学意义(F=16.646,P<0.01)。对照组分别与0.1 μmol/L 组(t=2.020,P>0.05)、0.5 μmol/L 组(t= 5.354,P>0.05)和1.0 μmol/L组(t=6.382,P<0.05)比较,与1.0 μmol/L实验组差异有统计学意义。4 组细胞形成的细胞集落大小无明显差异(图3)。
2.4 细胞免疫化学染色结果细胞免疫化学染色结果显示,p15ink4b蛋白和p16ink4a/CDKN2 蛋白阳性染色位于细胞浆,呈黄或棕黄色颗粒,未经处理的细胞胞浆无或略呈疏黄染颗粒,其大小形态较一致,核浆比较大。0.1 μmol/L 组、0.5 μmol/L 组、1.0 μmol/L 组细胞与对照组比较,细胞胞浆染色明显增强,呈黄色或棕黄色,阳性细胞增多,部分胞体变大,核仁清楚,胞浆增多,核浆比变小(图4、5,箭头所指)。
3 讨论

图4 IHC 检测5-Aza-CdR 对RKO 细胞p16ink4a/CDKN2 蛋白表达的影响(SP×200)Fig.4 p16ink4a/CDKN2 protein expression of RKO by Immunochemistry(SP×200)
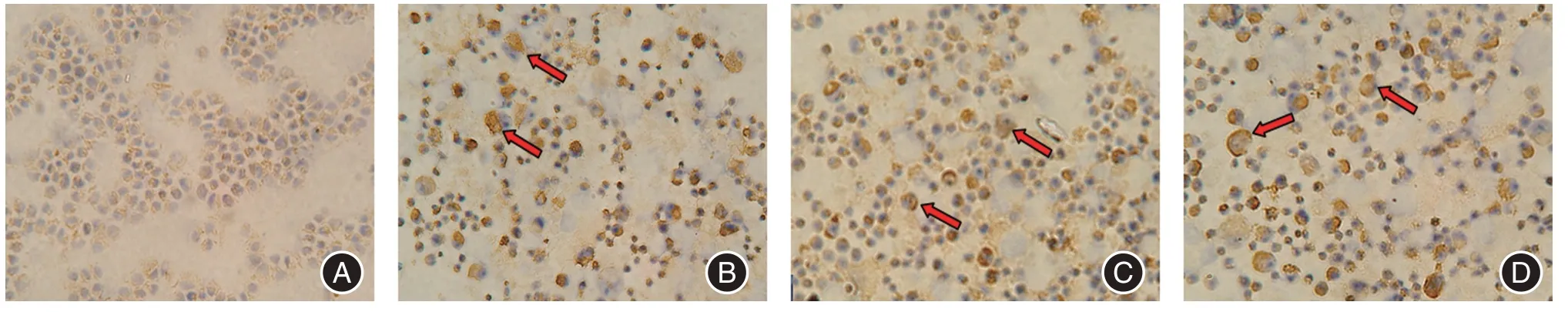
图5 IHC 检测5-Aza-CdR 对RKO 细胞p15ink4b蛋白表达的影响(SP×200)Fig.5 p15ink4b CDKN2 protein expression of RKO by Immunochemistry(SP×200)
已有研究表明,某些肿瘤抑制基因其DNA 序列完整,并未有突变、缺失等变化,无法解释其功能的失活,基于对染色体结构表遗传调控的再认识,认为碱基胞嘧啶环甲基化是其沉默原因,且DNA 甲基化贯穿于细胞癌变的各个阶段,是基因表达调控方式之一[1,6-7]。DNA 甲基化是指在DNMTs 催化作用下,由S-腺苷甲硫胺酸(S-adenosylmethionine,SAM)提供甲基,在CpG 二核苷酸5′-端胞嘧啶转变成5′-甲基胞嘧啶的过程。 近年来的研究认为[8-9],基因启动子区的甲基化与癌症、老年痴呆症的发生具有密切联系,而这种调控的关键在于DNMTs 的活性。DNMTs 呈高表达存在于多种肿瘤中[10-11],明显高于增生细胞,更高于正常细胞,其活性增高使抑癌基因的CpG 岛DNA 甲基化异常,使该基因无法正常表达,从而促使肿瘤增殖。本研究MTT 实验显示,给予不同浓度特异性DNMTs 抑制剂5-Aza-CdR 作用于肠癌RKO 细胞后,肠癌细胞生长活性均有显著抑制,并与5-Aza-CdR 浓度呈现明显的量效关系,说明DNMTs 的活性与肠癌细胞的分裂生长有关,与徐银辉等[12]报道的5-氮杂胞苷通过抑制DNMTs 的活性而使肺癌细胞生长增殖功能减弱甚至丧失结果相似。
软琼脂集落形成试验是测定单个细胞增殖能力的有效方法之一,其基本原理是单个细胞在体外持续增殖后,所组成的细胞群体成为克隆或集落,通过计数克隆形成率,可对单个细胞的增殖潜力作出半定量分析,克隆形成率高者其对环境的适应性及独立生存能力强,这也多少反映细胞恶性程度的特征。本实验结果显示3 个5-Aza-CdR干预组RKO 细胞集落数均显著少于对照组,并与5-Aza-CdR 剂量呈明显的抑制量效关系;同样的细胞迁移和侵袭实验显示肠癌细胞经5-Aza-CdR 作用后迁移数较对照组显著降低,两实验呈现出相近的结果,均提示5-Aza-CdR 能有效抑制肠癌细胞的迁移和侵袭力,并与5-Aza-CdR 浓度呈正相关;LI 等[13]报道5-Aza-CdR 通过下调Wnt 信号通路抑制肠癌干细胞的转移和生长;而MAIURI 等[14]则通过动物实验也显示相近结果,他们在小鼠结肠肿瘤模型中发现应用DNMTs 抑制剂能有效降低肿瘤的致瘤性。
本研究进一步探讨了5-Aza-CdR 对肠癌细胞生长增殖抑制作用的可能相关机制。我们选择了一类重要的细胞周期调控因子----细胞增殖抑制因子INK4(Inhibitor of CDK4)家族基因p15ink4b、p16ink4a/CDKN2 作为实验对象,它是特异性针对细胞周期素依赖激酶(cyclin-dependent kinasds)CDK4及CDK6 的肿瘤抑制基因,参与细胞周期的负调控,可防止细胞过度增殖和恶变[15],在前期实验已证实,该基因启动子区呈高甲基化,且这种异常的高甲基化可被5-Aza-CdR 逆转[4]。在本研究应用细胞免疫组化方法显示,通过5-Aza-CdR 作用后肠癌p15ink4b、p16ink4a/CDKN2 蛋白均进一步激活,表现为细胞胞浆染色黄色或棕黄色,表达增强,阳性细胞增多,部分胞体变大,核仁清楚,胞浆增多,核浆比变小,显现出一定的疗效关系,由此推究,肠癌细胞的抑制效应可能与5-Aza-CdR 逆转p15ink4b及p16ink4a/CDKN2 基因启动子区异常高甲基化从而激活蛋白表达、参与细胞负调控相关。因INK4 家族基因(p15ink4b及p16ink4a/CDKN2)为细胞周期G1/S 限制点负相关调控因子之一,即可抑制细胞从G1 进入S 期[16],也间接推断5-Aza-CdR 与细胞周期调节有关,国外学者已作类似结论[17],这点仍需实验进一步支持。
综上所述,肠癌细胞的发生发展机制较为复杂,参与的基因众多,具体是何种敏感性高特异性基因异常甲基化导致肠癌细胞的发生发展尚不清楚,DNMTs 的高表达使众多的抑癌基因失活沉默,但同时存在着许多癌基因表达增强,也有可能与肿瘤细胞的凋亡功能减弱相关[18-19]。本研究表明5-Aza-CdR 作为DNMTs 抑制剂的确具有抑制肠癌细胞的生长、增殖和侵袭能力,p15INK4b及p16ink4a/CDKN2 基因表达的激活可能是其机制之一,但对其他抑癌基因/癌基因影响如何仍待进一步研究;另外,本研究只是进行了细胞的体外实验,缺乏体内实验的有力支持,下一步值得探讨5-Aza-CdR对肠癌的动物模型的疗效及预后,以及联合其他化疗药物治疗的有效性。
