浅谈高效过滤器完整性测试在药包材和辅料洁净环境检验检测中的作用
2020-06-29王妮

摘 要:药包材和辅料是药品不可分割的重要辅助成分,与药品质量安全息息相关。部分直接接触药品的药包材是在C级或更高级别的洁净室内生产的,此时高效过滤器的完整性(泄漏率)对洁净环境的影响至关重要,因此安装完成和运行期间的完整性测试就成为极端必要的确认项目。现针对高效过滤器完整性测试,分析该项目在药包材和辅料洁净环境检验检测中的重要性和缺失原因,并由点及面,指出当前药包材和辅料生产企业在质量风险管理上的不足之处,有助于药包材和辅料生产企业加强质量风险管理的意识,协同制药企业提升药品质量,将药品质量风险降到最低。
关键词:高效过滤器;完整性测试;质量源于设计;质量风险管理
0 引言
药包材全称“直接接触药品的包装材料和容器”;辅料系指生产药品和调配处方时使用的赋形剂和附加剂,是除活性成分以外,在安全性方面已进行了合理的评估,且包含在药物制剂中的物质。药包材和辅料均是可能影响药品质量、安全性和有效性的重要组成部分,二者洁净厂房的环境检测项目和标准应当与制药企业一致。
医药行业所使用的高效过滤器一般是指对粒径在0.3 μm的最易穿透粒径(MPPS)粒子的捕集效率在99.99%~99.995%的过滤器[1],通常作为生产企业洁净车间的末端过滤装置,用以提供洁净的空气。自2011年《药品生产质量管理规范》实施以来,高效过滤器完整性测试一直是制药企业运行确认和回顾性验证的必检项目,但在药包材和辅料洁净车间的应用很少,在上海,仅一家包材生产企业(西氏)对洁净厂房高效过滤器完整性进行年度测试和确认(还有几家企业制袋和药液灌装共用一个生产车间,均按照《药品生产质量管理规范》进行管理,例如百特、海尼、长征富民)。
本文将简述高效过滤器完整性测试在药包材和辅料洁净环境检验检测中的重要性和缺失原因,并提出质量风险管理(Quality Risk Management)和质量源于设计(Quality by Design,QbD)理念应贯彻于药包材和辅料生产全过程。
1 高效过滤器完整性测试对药包材和辅料洁净环境检验检测的重要性
在国外,药品、医疗器械、药包材和辅料有一个统称,即医疗用品(medical products),因此药包材和辅料洁净环境的相关法规和标准均与药品相一致。美国《Sterile Drug Products Produced by Aseptic Processing — Current Good Manufacturing Practice》(2004版)第四章《厂房和设施》、欧盟《EU Guidelines to Good Manufacturing Practice Medicinal Products for Human and Veterinary Use》(附录1 无菌药品生产)(2008版)等国外法规标准都有对安装在洁净区的高效过滤器进行完整性测试的相关表述和接受标准。然而,在国内,药包材洁净室的高效过滤器完整性测试方法标准虽已确立,但尚未建立判断标准。《药品包装材料生产厂房洁净室(区)的测试方法》(YBB 00412004—2015)在“验证”这段有相关表述:“验证工作贯穿整个过程,包括施工前期设计、工程准备及承包商的选择以及整个施工周期的监控、项目竣工后静态运行阶段的测试(包括高效过滤器的检漏试验和自净时间等)[2]。”方法标准《洁净室施工及验收规范》(GB 50591—2010)、《洁净室及相关受控环境 检测技术分析与应用》(GB/T 36066—2018)以及ISO 14644-3主要对高效过滤器完整性测试方法进行了详细的描述。
高效过滤器完整性测试是指高效过滤器及其系统安装后的现场检漏,具体操作方法是采用气溶胶发生仪将其产生的多分散系气溶胶(一般为PAO,即poly alpha olefin)注入过滤器上游,使用光电扫描仪或特定的粒子计数器检测滤器下游的气溶胶浓度或计数值来判定滤器是否有泄漏,一般滤器上游的气溶胶浓度至少为10 μg/L以上。检测目的主要是检查过滤器滤材中的小针孔和其他零部件是否有损坏,如框架密封性、垫圈密封性以及过滤器构架是否存在漏缝等。对药包材和辅料企业来说,洁净室能否达到和保持设计的洁净级别,在一定程度上与高效过滤器的性能及其安装密切相关。因此,对高效过滤器进行完整性验证,确保其符合要求,是保证药包材和辅料生产车间洁净环境的重要手段之一。
2 高效過滤器完整性测试在药包材和辅料洁净车间管理中缺失原因
洁净生产的目的是控制洁净生产区域的悬浮粒子和微生物,而高效过滤器是洁净室环境控制最重要的支持部件,若未及时对高效过滤器的完整性进行检测,洁净生产环境的过程控制将会出现数据链断裂,无法做到预警,极大可能直接导致产品受到污染和交叉污染,使得产品发生系统性风险,即出现整批报废等情况。
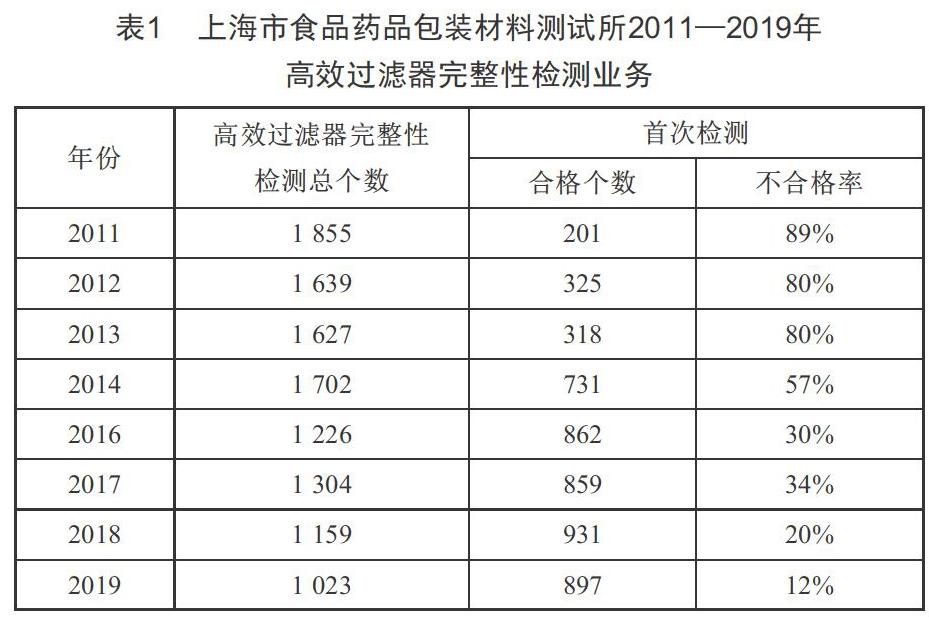
自2011年《药品生产质量管理规范》实施以来,我所每年为制药企业进行高效过滤器完整性检测约25家次,超过1 000个高效过滤器。其中,2011年刚开始执行该法规的时候首次检测高效过滤器不合格的比例高达89%,随后每年的检测不合格率下降趋势明显,直至2019年,首次检测高效过滤器不合格的比例已降低至12%,如图1、表1所示。可见,法规层面的落实和定期对高效过滤器实施确认和验证有助于生产过程控制和风险排查,可将各类风险隐患消除在萌芽阶段,以防控重大质量安全事故。
高效过滤器完整性测试在药包材和辅料洁净车间的应用非常少,导致该现象产生的原因,一方面是《直接接触药品的包装材料和容器管理办法》(局令第13号)和《药品包装材料生产厂房洁净室(区)的测试方法》(YBB 00412004—2015)中关于验证和确认的目的仅是确认厂房和设施设备等的正常运行,确保能维持正常生产,并未对高效过滤器完整性标准有具体的规定,标准中对于确认与验证的表述其可操作性和重要性仍然不足,致使药包材和辅料生产企业对该项目的重视度和认知度不高。但《药品生产质量管理规范》(2010年修订)和国外的相关法规标准已把验证和确认作为GMP的核心和灵魂所在,高效过滤器完整性测试应成为药包材和辅料洁净厂房确认和验证的一个重要组成部分。
另一方面是药包材和辅料生产企业对于QbD理念的重视程度与制药企业相差甚远。QbD首次出现在人用药品注册要求国际协调会(ICH)Q8,是一种系统的研发方法,其以预先设定目标为起始,基于可靠的科学和质量风险管理,强调对产品和生产过程的理解及对工艺的控制[3]。这种以始为终、先设定目标再行动的创新管理模式现已在药品研发和工艺设计方面应用广泛,正逐渐渗透于药品生产全过程的管理中。其中,洁净生产环境是药品生产全过程控制的一个重要组成部分。制药企业在厂房建造前,会根据产品类型、生产效率、生产管理、产品过程控制和检验检测管理,进行合适的空间平面布局设计、合理的人流物流设计、便于检验检测的预留设计及合适的建筑装修材料的选择[4]。洁净厂房若要进行高效过滤器完整性测试,必须在厂房设计时,预留上游浓度测试孔,以检测到上游浓度,同时还必须对高效过滤器的材质和施工工艺提出更高的要求。目前QbD理念仅停留在制药企业,如何将QbD理念深刻植入药包材和辅料生产企业洁净车间的设计和管理中,把QbD理念结合过程分析技术(PAT)覆盖到药包材和辅料产品由开发开始的整个生命周期中,仍是一个亟需解决的严峻问题。
3 质量风险管理的定义及其在药品生产全过程中的重要性
质量风险管理最初是由美国食品药品监督管理局(FDA)在2002年引入制药行业。ICH紧随其后,在2005年发布了Q9。2010年,中国国家食品药品监督管理局(SFDA)发布了《药品生产质量管理规范》(2010年修订),将药品质量风险管理写入其中[1],其表述为:“质量风险管理是在整个产品生命周期中采用前瞻或回顾的方式,对质量风险进行评估、控制、沟通、审核的系统过程” “应当根据科学知识及经验对质量风险进行评估,以保证产品质量” “质量风险管理过程所采用的方法、措施、形式及形成的文件应当与存在风险的级别相适应”[5]。
增加质量风险管理的标准,体现了立法者深刻理解药品安全的本质是控制药品风险。国家要求制药企业应建立质量风险管理体系,以提升风险管理能力,进而规避风险,为患者提供安全可靠的药品产品。目前,随着国家对药品监管的力度不断加强,国内多数制药企业的风险管理能力和质量意识已有所提高,能够通过完善企业质量风险管理体系,满足药监部门审查要求,进而保障企业规范化生产。
然而,绝大多数的药包材和辅料生产企业的风险管理体系和风险管理机制至今仍处于未设立状态或萌芽阶段。2015年,《药品包装材料生产厂房洁净室(区)的测试方法》(YBB 00412004—2015)在洁净室环境的测试方法上已与《药品生产质量管理规范》(2010年修订)接轨,把风险管理的内容结合到测试项目中,提出对相关检测数据作分类整理和趋势分析,用以判断空调净化系统的性能和开展风险评估[2]。但该标准由于缺少相关指南,没有可操作性。2017年,中国成功加入ICH,标志着由ICH颁布的一系列技术指南将成为我国药品监管的技术标准,药品生产的质量风险管理将同步接轨ICH Q9。药包材和辅料对保证药品质量和保障人体用药安全具有重要作用,其生产过程的质量风险管理也应得到广泛关注和重视。
4 结语
制药行业与人的生命直接相关,药包材和辅料产品均是药品生产全过程中的重要因素,可能存在着影响药品质量和患者用药安全的风险。因此,制药企业应携同药包材和辅料生产企业严格实施质量风险管理,以使整个药品生产全过程始终处于受控状态,抓紧每个生产环节,进而保证药品质量,杜绝安全隐患,降低質量风险。
立足洁净室检验机构的角度,希望药包材和辅料洁净环境检测尽快与制药企业保持一致和同步,对标国际法规和技术指南,用法律法规、技术标准加强生产环境的日常监控、确认和验证。此外,药包材和辅料生产企业应贯彻QbD理念,在厂房设计中将高效过滤器完整性测试等检测和管理纳入考虑范围,增强定期确认和验证的意识,以保证产品安全、合规生产。
[参考文献]
[1] 敖唯一,关元宙,梁毅.基于中药丸剂生产工艺的质量风险管理[J].中国医药工业杂志,2019,50(8):943-948.
[2] 国家药典委员会.国家药包材标准[S].北京:中国医药科技出版社,2015.
[3] 国家食品药品监督管理局药品认证管理中心.欧盟药品GMP指南[M].北京:中国医药科技出版社,2008.
[4] 李正.醋酸曲安奈德注射液生产线工程设计的风险管理研究[D].上海:华东理工大学,2016.
[5] 药品生产质量管理规范(2010年修订):中华人民共和国卫生部令第79号[A],2011.
收稿日期:2020-04-01
作者简介:王妮(1993—),女,上海人,助理工程师,研究方向:药包材和辅料相容性、洁净空气净化系统验证。
