基于网络药理学的大黄抗炎作用机制分析*
2020-06-12仁增加切羊让忠华吉卓玛贡却坚赞
仁增加,切羊让忠,2,华吉卓玛,贡却坚赞,2※
(1.青海大学藏医学院,青海 西宁 810016;2.藏药新药开发国家重点实验室,青海 西宁 810016;3.海西州蒙藏医医院,青海 德令哈 817000)
大黄为寥科植物掌叶大黄palmatumL.、唐古特大黄RheumtanguticumMaxim,exBalf.或药用大黄RheumofficinaleBaill.的干燥根和根茎,是中藏药常用的药材。现代研究表明,大黄成分主要包括蒽醌类衍生物、蒽酮类衍生物和二苯乙烯类、鞣质类、酰基糖苷类、色酮类、苯丁酮苷类等各类型化合物,具有泻下、抗肿瘤、抗炎镇痛、保肝利胆、降血脂、改善肾功能等药理作用。近几年,有关大黄抑制炎症发生发展的研究报道较多[4],但从细胞和分子层面揭示大黄抗炎作用机制的研究甚少。网络药理学是基于系统生物学的理论,对药物的有效成分进行多靶点、多途径生物系统网络图分析的学科。本研究采用网络药理学方法对大黄有效成分的抗炎关键靶点、功能、通路进行分析,揭示其抗炎作用机制,为后续研究提供参考。
1 材料与方法
1.1 化学成分查找与主要活性成分筛选
本文采用中药系统药理学数据库与分析平台TCMSP(http://lsp.nwu.edu.cn/tcmsp.php)检索大黄的所有化学成分,共查找了92个化合物。通过评价成分的体内过程,以口服生物利用度(oralbioavailability,OB)≥30%和类药性(drug-like,DL)≥0.18为标准筛选中药的活性成分[1]。其中OB值是评价药物能否发挥药效的重要指标,DL值代表成分与已知化学药的相似性,这两个值对确定中药成分是否对机体产生活性具有重要参考价值[2]。
1.2 活性成分对应的靶点信息获取
根据筛选出来的活性成分在TCMSP数据库中查找与之相对应的靶点,并进一步使用R软件将其靶点数据在UniProt数据库进行靶基因注释及确认。运用Cytoscape 3.7.2软件(http://www.Cytoscape.org/)构建大黄化合物-靶点网络图。
1.3 疾病靶点的收集与整理
以inflammation、inflammatory为检索词,在人类基因数据库Genecards(https://www.genecards.org/)、人类孟德尔遗传数据库OMIM(https://omim.org/)检索与之相关的靶点,将Relevance score设为大于5,且应用Venny 2.1.0在线软件筛选大黄和炎症的交集靶点并绘制韦恩图。
1.4 蛋白互作网络(PPI)的构建与分析
将大黄和炎症的交集靶点上传至String数据库(https://string-db.org/),物种选为Homo sapiens,最低要求的评分条件设定为中等置信度0.4,通过高级设置隐藏网络图中游离的蛋白质,其余参数设置为默认,再将TSV文本数据导入cytoscape 3.7.2软件绘制蛋白互作网络图。
1.5 GO功能和KEGG通路富集分析
将获取的关键靶点导入Metascape数据库,设置参数如下,物种选“H.sapinens”;Min Overlap:3,P Value:0.01,Min Enrichment:1.5;GO生物功能选GO Molecular Functions、GO Biological Processes、GO Cellular Components,Pathway选Canonical Pathways、Reactome Gene Sets、KEGG Pathway数据库。然后进行GO生物功能和KEGG通路富集分析,并运用KEGG mapper工具绘制主要通路的通路图。
2 结果与分析
2.1 大黄活性成分分析
在TCMSP中将OB≥30%、DL≥0.18设为筛选条件,并补充常用组分,剔除非人员靶标,最终获得7个有效活性成分,详见表1。我们发现芦荟大黄素具有很高的口服利用度,而β-谷甾醇具有很高的类药性。
2.2 化合物-靶点网络构建
从TCMSP数据库中共检索到与大黄活性成分相对应的靶基因74个,并使用Uniport数据库对靶基因进行注释及确认,运用Cytoscape 3.7.2软件将大黄的7个化合物和74个靶点进行映射,构建化合物-靶点网络图,见图1。图中内圈为化合物,中圈(degree=1)和外圈(degree≧2)为靶点,节点大小表示度值的大小。其中beta-sitosterol(degree=26)、aloe-emodin(degree=19)是化合物中度值最高的,靶点NCOA2和PTGS2的度值为6,度值越大,表明其抗炎的贡献越高。
表1 大黄的活性成分及OB和DL值

Table 1 The Active Ingredients of Rhubarb and the values of OB&DL

图1 化合物-靶点网络图
Figure 1 The Network Diagram of Compound-Target Spot
2.3 大黄与炎症的交集基因计算
从Genecards和OMIM数据库中检索与炎症相关的所有靶点,设置筛选条件,相关性得分大于5后得1 208个靶点,运用Venny 2.1.0软件绘制大黄与炎症的韦恩图得到25个交集基因,见图2。

图2 大黄与炎症的交集基因韦恩图
Figure 2 The Venn Diagram of Intersection Gene Between Rhubarb and Inflammation
2.4 大黄PPI互作网络的构建与分析
为更进一步分析大黄治疗炎症的作用机制,将25个交集基因上传至String数据库获取TSV文本数据,利用Cytosccape 3.7.2软件绘制蛋白互作网络图,见图3。该网络中25个靶点相互作用产生了85条蛋白互作的边,平均度值为7.39,平均介数为0.039,根据节点的度值和介数中心性筛选出12个靶点,见表2。推测这些靶点可能是大黄抗炎的关键靶点,其中MYC与细胞周期、凋亡、生长及血管生成有关,CASP3、JUN、CASP8、AR等与细胞凋亡有关,PTGS2、PGR与炎症有关,IL1B与炎症和细胞增殖、分化、凋亡相关,CDKN1A与细胞周期有关,PPARG跟血管调节有关,ESR1与细胞增殖相关,KDR与细胞增殖、存活、迁移有关,从以上靶点功能可预测,大黄可能都是通过细胞增殖、凋亡和血管调节发挥抗炎作用。

图3 大黄PPI蛋白互作网络图
Figure 3 Network Diagram of the Rhubarb and PPI Protein Interaction
表2 关键靶点拓扑学参数

Table 2 The Topological Parameters of Key Target Spot
2.5 GO功能富集及KEGG通路富集分析
利用Metascape平台对大黄的关键靶点进行GO功能富集分析,P设为0.01,获取其参与生物过程的前20个基因功能富集,见图4,主要涉及类固醇激素反应(11个靶点)、凋亡信号通路(12个靶点)、有毒物质响应(11)、细胞程序性死亡的正调控(11个靶点)、营养水平的反应(10个靶点)、脂多糖的反应(8个靶点)、MAPK级联的调节(9个靶点)、细胞内信号转导的负调控(8个靶点)、氧含量的反应(7个靶点)、生长增殖的负调控(6个靶点)、损害的反应(8个靶点)、转运蛋白活性的调节(5个靶点)、活性氧代谢过程的调节(5个靶点)等,表明大黄参与多种生物过程调控抗炎的作用机制。

图4 GO功能富集分析柱状图
Figure 4 The Histogram of Analyzing GO Functional Enrichment
通过KEGG通路富集分析发现,P小于0.01的信号通路有13条,分别涉及癌症通路(13个靶点)、核受体转录途径(6个靶点)、炎症并发症中的AGE-RAGE信号通路(6个靶点)、PID ERA基因组途径(5个靶点)、IL-17信号通路(5个靶点)、PID NFAT TF通路(4个靶点)、PID RXR VDR通路(3个靶点)、5-羟色胺能突触信号通路(4个靶点)、MAPK信号通路(5个靶点)、PID P53下游通路(4个靶点)、流体剪切应力和动脉粥样硬化通路(4个靶点)等,见图5。并用KEGG mapper工具绘制癌症通路的通路图。上述结果提示大黄可能通过以上多条信号通路发挥抗炎作用。

图5 KEGG通路富集分析网络图
Figure 5 The Network Diagram of Pathways Enrichment of KEGG Analysis
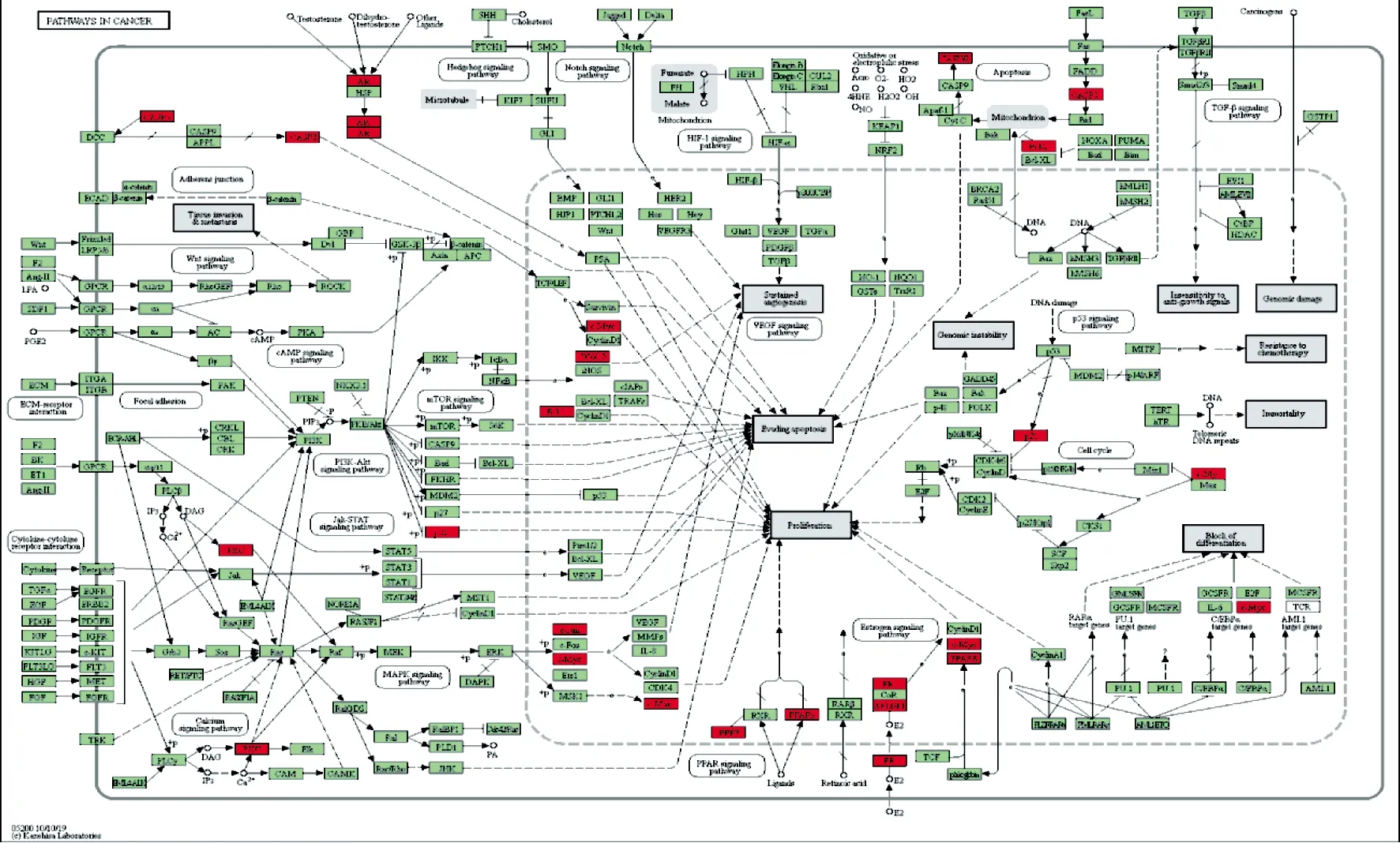
图6 癌症信号通路图
3 讨论
已有研究报告显示大黄具有抗炎的药理作用,但是它们发挥作用的全面机制以及其他成分在抗炎中的作用机制还不完全清楚。大黄作为一种常见的中藏药,其成分复杂,具有中药典型的多成分、多靶点特点。因此,本研究利用网络药理学的方法,并结合其他相关数据库和软件对大黄治疗炎症的可能机制进行了探讨。研究发现大黄中发挥作用的主要有效成分有7个,其中芦荟大黄素具有很高的口服利用度,而β-谷甾醇具有很高的类药性。从TCMSP数据库中共检索到大黄对应靶点74个,其中有效靶点25个,β-谷甾醇和芦荟大黄素分别有26、19个靶点,β-谷甾醇是通过抑制炎症因子IL-1β、IL-6、MCP-1等和相关酶的过表达缓解急性结肠炎[3]。李晓红等[4]和Hu等[5]考察芦荟大黄素的抗炎作用及机制,结果显示,芦荟大黄素通过抑制诱导型一氧化氮合酶(i NOS)mRNA 表达,可明显降低脂多糖刺激RAW264.7细胞的一氧化氮释放,从而发挥抗炎作用。
根据PPI互作网络结果显示,MYC、Casp3、JUN、PTGS2、IL1β、ESR1等是大黄抗炎的主要靶点,如MYC是一个强大的原癌基因,有c-MYC、N-MYC、L-MYC三种[6],c-MYC在细胞增殖和凋亡中具有双重作用,与炎症组织损伤和修复的多个方面有关[7]。Casp3调节不同的生物过程如凋亡、炎症等[8]。JUN 是哺乳动物细胞内AP-1亚单位之一,而 AP-1是位于JNK下游的关键信号蛋白分子,JNK受刺激后,可激活JUN,使其磷酸化,从而使炎症转录启动并增强,导致炎症持续和发展[9]。PTGS2 是前列腺素合成过程中的关键酶,而前列腺素是一种重要的炎症介质,参与机体炎症反应[10]。白介素-1β介导小胶质细胞释放细胞因子,刺激单核-巨噬细胞产生TNF-α,促进内皮细胞表达黏附分子,加重局部炎症反应[11]。ESR1可调节脂肪组织的血管内皮生长因子A(VEGFA),增强血管生成,减少炎症和改善脂肪组织功能[12]。
GO功能富集分析结果显示,GO功能富集主要涉及类固醇激素反应、营养水平的反应、脂多糖的反应、氧含量的反应、损伤反应、有毒物质响应、凋亡信号通路、细胞程序性死亡的正调控、细胞内信号转导的负调控、MAPK级联的调节、转运蛋白活性的调节、生长增殖的负调控、活性氧代谢过程的调节。KEGG通路分析结果显示,KEGG通路分别涉及癌症通路、炎症并发症中的AGE-RAGE信号通路、IL-17信号通路、PID NFAT TF通路、PID RXR VDR通路、MAPK信号通路、PID P53下游通路、PID ERA基因组途径和核受体转录途径通路、5-羟色胺能突触信号通路、流体剪切应力和动脉粥样硬化通路等。
综上,本研究运用网络药理学方法从分子生物学层面分析了大黄的多成分、多靶点协同作用的抗炎机制。本研究结果为进一步深入研究大黄抗炎机制奠定了基础。
