一种前列腺特异性膜抗原小分子抑制剂的合成新路线
2020-06-11肖清炜常媛媛刘起发臧小豪胡蒙蒙
肖清炜,常媛媛,刘起发,臧小豪,胡蒙蒙,周 渭
(广东工业大学 生物医药学院,广东 广州 510006)
前列腺癌(Prostate Cancer, PCa)是男性泌尿生殖系统最常见的恶性肿瘤,占据男性癌症整体发病率第二位[1]。其中,发达国家的男性发病率要高于发展中国家[2]。但近年来,随着医疗手段的进步与生活环境的变化,包括中国在内的发展中国家中男性前列腺癌的发病率也呈逐年上升趋势[3]。针对癌症的治疗,早期诊断与精准治疗是提高癌症患者的生存率与治愈率的良好手段。 因此,研究与开发出一种前列腺癌的早期诊断治疗手段是当前的研究热点。
分子探针技术的开发极大地提高了对疾病的诊断效果[4]。其原理是利用某种疾病的特异性靶点开发出一种靶向性的分子影像探针从而达到诊断与治疗的目的。因此,前列腺癌的早期诊断也可通过分子探针技术手段实现。其中,前列腺特异性膜抗原(Prostate specific membrane antigen, PSMA)作为前列腺癌的特异性靶点越来越受到研究者的关注[5]。
PSMA是一种表达在前列腺肿瘤上皮细胞,含有750个氨基酸的II型跨膜糖蛋白[6-7],其与II型谷氨酸羧肽酶(GCPII)具有相同的酶活性。II型谷氨酸羧肽酶是存在于神经系统中用于催化水解Nacetylaspartylglutamate(NAAG)释放谷氨酸的一种膜外金属蛋白酶。但对于大脑神经系统,过量的谷氨酸会导致一系列的神经性疾病,包括中风、脊髓损伤、肌萎缩侧索硬化(ALS)、周围神经病变、慢性疼痛、精神分裂症和癫痫[8]。抑制GCPII活性将有利于治疗由谷氨酸释放过多所引发的神经性疾病。因此,近年来,针对II型谷氨酸羧肽酶的抑制剂得以被研究与开发。如图1所示,Cyril Barinka和Youngjoo Byun等[9]基于GCPII所作用的NAAG结构通过理论计算设计合成了含有谷氨酸-脲(Glu-Urea)骨架的GCPII型小分子抑制剂并通过多种评价手段对4种抑制剂的结合机理与结合效果进行了评价,实验结果证明所设计合成的Glu-Urea骨架小分子抑制剂能够与GCPII特异性结合。同时,因为PSMA与GCPII具有相同酶活性,因此所开发的小分子抑制剂也能与PSMA进行特异性结合,这给前列腺癌靶向性分子的开发提供了一种新的途径[7,10]。当前,已经有不少研究利用PSMA的小分子抑制剂进行靶向分子探针或药物的开发[10-14]。因此,设计出一条合适的PSMA小分子抑制剂及其类似物的全合成路线对促进前列腺癌靶向性分子影像探针及药物的开发有着重要的意义。
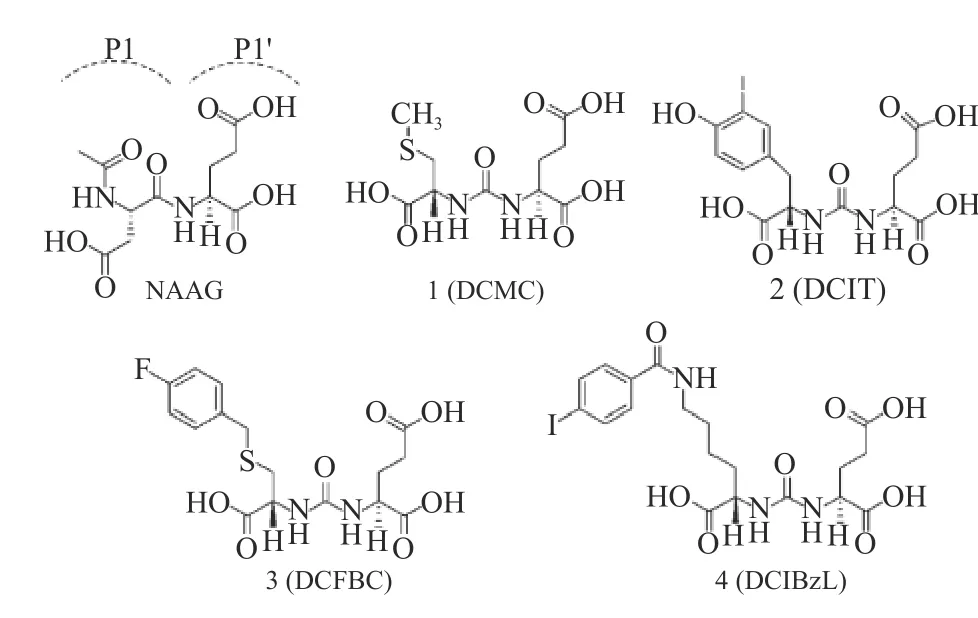
图 1 NAAG及4种具有Glu-Urea骨架的GCPII型小分子抑制剂分子结构[9]Fig.1 The molecular structure of NAAG and Four GCPII Small Molecular Inhibitors with Glu-Urea Framework
PSMA小分子抑制剂的合成路线已见诸报道,基本策略是以L-谷氨酸二叔丁基酯为起点,通过与其他氨基酸的氨基反应构成谷氨酸-脲骨架结构。例如,陈骁驰等[15]就提供了一种通过L-谷氨酸叔丁基二酯与苄氧羰基(Cbz)和叔丁氧羰基(Boc)保护的L-赖氨酸通过三光气反应得到了具有Glu-Urea-Lys骨架的PSMA小分子抑制剂。但到目前为止,还没有对以L-谷氨酸和L-赖氨酸为起始原料的合成路线的研究报道。因此,基于PSMA的小分子抑制剂及其类似物在前列腺癌分子影像学诊断及靶向治疗中的作用,笔者提供了一种以L-谷氨酸和L-赖氨酸为起始原料的合成路线,为利用PSMA为靶点的前列腺癌的诊断与治疗手段提供小分子抑制剂的合成策略。
1 实验部分
1.1 实验仪器与试剂
DF-101D型集热式磁力搅拌器(郑州予华);DHJF-2005低温搅拌反应浴(郑州长城科工贸);AVANCE III HD 400型核磁共振波谱仪(瑞士布鲁克);Q Exactive型超高分辨液质联用仪(Thermo-Filsher公司);MCP 500自动旋光仪(德国安东帕公司)。
L-谷氨酸、L-赖氨酸等所有药品均购自阿拉丁化学试剂公司,所有化学试剂均为分析纯。
1.2 实验方法
如图2所示,PSMA小分子抑制剂的合成将按以下路线进行,化合物1、2、3 ···与图中所进行编号的化合物一一对应。
(1) L-谷氨酸二叔丁基酯(化合物1)的合成。
取5 g L-谷氨酸溶于100 ml的醋酸叔丁酯中并置于冰浴中搅拌5 min后,缓慢滴加2.0 eqv的70%高氯酸溶液,置于室温搅拌过夜。反应终了后,将反应液降至0 ℃,并用冷的0.5 mol/L盐酸(2×30ml)萃取,收集水相。再将碳酸钠固体缓慢加入至水相中并搅拌至pH为8~10,二氯甲烷萃取,无水硫酸钠干燥。过滤后的液体减压旋蒸得到目标化合物1,无色或浅黄色透明黏性液体。产率:47.6%,1H NMR (400 MHz,CDCl3) δ :3.50~3.40 (m, 1H), 3.03 (s, 3H), 2.37 (t, J=7.5 Hz,2H), 2.02 (d, J=10.3 Hz, 1H), 1.90~1.76 (m, 1H), 1.47(t, J=8.9 Hz, 18H)。13C NMR (101 MHz, CDCl3)δ:173.95, 172.61, 81.73, 80.64, 54.11, 31.82, 29.44,28.09, 28.03。HR-MS(ESI)m/z: Calcd for C13H25NO4{[M+H]+}260.18524, found 260.18563,[α]D20=22.86 (C=0.687,CH3OH)。
(2) N6-(9 -芴甲氧羰基)-N2-叔丁氧羰基-L-赖氨酸(化合物2)的合成。
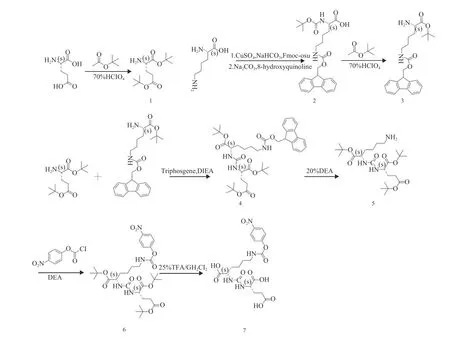
图 2 小分子抑制剂PSMA-1的合成路线Fig.2 The synthesis route of small molecule inhibitors PSMA-1
化合物2的合成分为两步骤进行。第一步:17 g碳酸氢钠加入到50 ml水中,搅拌形成碱液,将10 g L-赖氨酸盐酸盐加入其中搅拌15 min。再分批向反应器中加入7 g无水硫酸铜搅拌1h,后加入22.1 g 9-芴甲基-N-琥珀酰亚胺碳酸酯和100 ml的四氢呋喃。搅拌14 h后加入100 ml碳酸氢钠水溶液搅拌3 h。过后加入50 ml水和20 ml甲醇搅拌5 h后,加入50 ml乙酸乙酯搅拌30 min。最后过滤,滤渣至水洗为无色。真空干燥箱烘干后得到N6-Fmoc-L-赖氨酸的铜络合物中间体。第二步:取烘干好的中间体10 g溶于四氢呋喃:水=1:1溶液中,依次分批加入10 g无水碳酸钠与5 g 8-羟基喹啉。室温搅拌2 h后往上述体系中加入6 g的二碳酸二叔丁酯(Boc酸酐)。继续室温搅拌5 h,过滤,滤液冷却至5 ℃以下。用盐酸调节pH至2~3,萃取得到粗产品,硅胶柱层析提纯得到目标化合物2,黄色粘稠状液体。两步骤合并总产率:65%,1H NMR (400 MHz,CDCl3) δ: 7.67 (t, J=6.9 Hz, 2H), 7.51 (d, J=7.2 Hz,2H), 7.32 (t, J=7.4 Hz, 2H), 7.23 (t, J=7.3 Hz, 2H), 6.08(s, 0.4H), 5.58 (s, 0.2H), 5.13 (s, 0.6H), 4.85 (s, 0.8H),4.27 (dd, J=62.5, 28.3 Hz, 6H), 3.11 (d, J=5.9 Hz, 2H),1.89~1.22 (m, 13H)。13C NMR (101 MHz, CDCl3) δ:176.46, 156.74, 155.88, 143.97, 127.70, 127.08, 125.09,119.98, 80.17, 66.70, 53.27, 47.27, 40.63, 32.05, 29.35,28.36, 22.44。HR-MS(ESI)m/z: Calcd for C26H32N2O6{[M-H]-}467.21884,found 467.21876。
(3) N6-(9 -芴甲氧羰基)-L-赖氨酸叔丁酯(化合物3)的合成。
取化合物2(1.0 eqv,2 g)溶于50 ml的醋酸叔丁酯中低温搅拌5 min,缓慢滴加1.5 eqv的70%高氯酸溶液,并置于室温搅拌过夜。反应终了后,将反应液冷却至0 ℃,小心加入2 mol/L的NaOH水溶液至pH为10。然后立即用乙酸乙酯萃取,无水硫酸钠干燥后过滤,硅胶柱层析提纯得到目标化合物3,无色透明黏性液体。产率70%,1H NMR (400 MHz, CDCl3) δ : 7.70 (d, J=7.5 Hz, 2H), 7.57 (d, J=7.3 Hz, 2H), 7.34 (t, J=7.4 Hz,2H), 7.26 (dd, J=9.9, 4.7 Hz, 2H), 4.39-4.24 (m, 1H),4.15 (d, J=8.3 Hz, 0H), 4.00 (t, J=5.9 Hz, 1H), 3.12 (s,1H), 1.94 (d, J=5.2 Hz, 1H), 1.46 (d, J=30.9 Hz, 14H)。13C NMR (101 MHz, CDCl3) δ : 171.28, 168.49,156.96, 143.99, 141.24, 127.66, 127.11, 125.26, 125.08,119.89, 84.69, 66.81, 60.45, 54.16, 47.12, 40.34, 29.97,29.13, 27.82, 21.72, 21.07, 14.21。HR-MS(ESI)m/z:Calcd for C25H32N2O4{[M+H]+}425.24310, found 425.24348, [α]20D=7.9(C=1,CH3OH)。
(4) N6-(9 -芴甲氧羰基)-N2-(N-甲酰基-L-谷氨酸二叔丁基酯)-L-赖氨酸叔丁酯(化合物4)的合成。
取化合物3(1.0 eqv,1 g),溶解在二氯甲烷溶液中,置于0 ℃冰浴搅拌5 min后加入三光气(0.4 eqv,0.28 g)并继续在上述条件下反应,TLC跟踪反应。反应1~2 h后加入化合物1(1.0 eqv,0.612 g)和二异丙基乙胺(DIEA)(4.0 eqv,1.22 g)冰浴搅拌10 min后移至室温搅拌至TLC检测反应完全。硅胶柱层析提纯(石油醚:乙酸乙酯=4:1)得到目标化合物4,冷却条件下为白色膏状固体,置于室温为无色透明粘稠状固体。产率:75.3%,1H NMR (400 MHz, CDCl3) δ : 7.75 (d,J=7.5 Hz, 2H), 7.61 (t, J=6.4 Hz, 2H), 7.39 (t, J=7.4 Hz,2H), 7.30 (td, J=7.4, 0.9 Hz, 2H), 5.30 (d, J=8.1 Hz, 3H),4.48~4.28 (m, 4H), 4.21(s, 1H), 3.18 (s, 2H), 2.28 (d,J=6.8 Hz, 2H), 2.04 (s, 1H), 1.91~1.79 (m, 1H),1.67~1.29(m, 32H)。13C NMR (101 MHz, CDCl3) δ:172.60, 172.49, 172.38, 156.98, 156.65, 144.07, 141.30,127.63, 127.04, 125.17, 119.92, 82.16, 81.73, 80.52,66.55, 53.35, 53.05, 47.33, 40.67, 32.69, 31.60, 29.34,28.33, 28.08, 28.04, 28.01, 22.37。HR-MS(ESI)m/z:Calcd for C39H55N3O9{[M+Na]+}732.38275, found 732.38305。
(5) N2-(N-甲酰基-L-谷氨酸二叔丁基酯)-L-赖氨酸叔丁酯(化合物5)的合成。
取2 g化合物4置于10%的二乙胺溶液中搅拌,TLC检测反应完全。减压旋蒸后柱层析提纯(乙酸乙酯:甲醇=25:1至1:1)得到目标化合物5,黄色黏性固体。产率:70%,1H NMR (400 MHz, MeOD) δ:4.02(ddd, J=16.9, 8.4, 5.1 Hz, 2H), 2.51 (t, J=7.1 Hz, 2H),2.26~2.02 (m, 2H), 1.88 (dd, J=7.8, 5.9 Hz, 1H), 1.62 (s,2H), 1.52~1.42 (m, 1H), 1.42~1.22 (m, 31H)。13C NMR(101 MHz, MeOD) δ : 172.53, 172.33, 172.08, 158.56,81.38, 81.15, 80.33, 53.41, 52.80, 40.72, 31.99, 31.32,31.13, 27.63, 27.02, 26.97, 26.94, 22.53。HRMS(ESI)m/z: Calcd for C24H45N3O7{[M+H]+}488.33295, found 488.33303。
(6) N6-(对硝基苯氧羰基)-N2-(N-甲酰基-L-谷氨酸二叔丁基酯)-L-赖氨酸叔丁酯(化合物6)的合成。
取化合物5(1.0 eqv,1 g)、三乙胺(1.0 eqv,0.21 g)溶于20 ml二氯甲烷溶液中,再加入对硝基苯基氯甲酸酯(1.2 eqv,0.49 g),室温下搅拌,TLC跟踪反应,反应结束后用减压旋蒸掉多余溶剂,硅胶柱层析提纯(石油醚:乙酸乙酯=2:1)得到目标化合物6,无色透明黏性液体。产率:73%,1H NMR (400 MHz, CDCl3) δ :8.288.20 (m, 2H), 7.39~7.30 (m, 2H), 6.01 (s, 1H), 5.29(d, J=8.1 Hz, 1H), 5.20 (s, 1H), 4.37 (qd, J=8.1, 4.8 Hz,2H), 3.27 (d, J=6.0 Hz,2H), 2.45~2.22 (m, 2H),2.15~2.02 (m, 1H), 1.95~1.72 (m, 2H), 1.65~1.54 (m,3H), 1.45 (dd, J=10.8, 7.6 Hz, 29H)。13C NMR (101 MHz, CDCl3) δ : 173.29, 172.37, 172.22, 157.25,156.33, 153.42, 144.52, 125.03, 121.89, 82.58, 81.80,80.67, 53.27, 53.01, 41.06, 32.72, 31.54, 28.81, 28.16,28.05, 28.01, 22.55。HR-MS(ESI)m/z: Calcd for C31H48N4O11{[M+H]+}675.32105, found, 675.32118。
(7) N6-(对硝基苯氧羰基)- N2-(N-甲酰基-L-谷氨酸)-L-赖氨酸(PSMA-1)的合成。
取1 g化合物6溶于含有25%TFA的二氯甲烷溶液中反应16 h,反应液经减压旋蒸掉多余溶液后硅胶柱层析提纯(甲醇:乙酸乙酯=1:1)得到目标化合物7,无色透明黏性固体。产率:85%,1H NMR (400 MHz,MeOD) δ : 8.27 (d, J=9.1 Hz, 2H), 7.37 (d, J=9.1 Hz,2H), 4.33 (ddd, J=13.2, 8.3, 5.0 Hz, 2H), 3.23 (t, J=6.8 Hz, 2H), 2.49~2.37 (m, 2H), 2.23~2.08 (m, 1H),2.00~1.81 (m, 2H), 1.71 (dd, J=14.0, 7.3 Hz, 1H), 1.62(dd, J=11.4, 7.0 Hz, 2H), 1.51 (dd, J=14.9, 7.5 Hz, 2H)。13C NMR (101 MHz, MeOD) δ: 175.21, 174.63, 158.74,156.36, 154.30, 144.70, 124.68, 122.00, 52.68, 52.21,40.47, 31.65, 29.76, 28.78, 27.55, 22.45。HRMS(ESI)m/z: Calcd for C19H24N4O11{[M-H]-}483.13697, found 483.13688, [α]20D=11.3(C=1,CH3OH)。
2 结果与讨论
本文以L-谷氨酸和L-赖氨酸等基础化合物为原料,共计7步反应得到具有Glu-Urea-Lys骨架的PSMA小分子抑制剂类似物。各步骤产物均进行了1H NMR、13C NMR及高分辨质谱表征,对化合物1,3与7进行了旋光测试证明其仍保持光学活性。各步骤产率除化合物1的合成外均高于60%,线路总产率为18%。两种初始中间体, 化合物1与化合物3的产率分别为47.6%与65%。伴随着目标产物的合成,笔者开发了一种针对L-赖氨酸羧基与ε氨基保护基保护的合成新思路。中间体化合物3的合成本质上是氨基酸活性基团的保护与脱保护。我们首先通过9-芴甲基氧羰基与叔丁氧羰基对L-赖氨酸ε氨基与α氨基进行了保护,合成得到了化合物2。相较于其他文献中ε氨基进行苄氧羰基保护的L-赖氨酸中间体,9-芴甲基的优点在于其脱保护时只需要二乙胺等有机胺即可短时间内脱除,对比苄氧羰基的Pt/C +H2脱除法,成本更低,实验更安全。其次,在合成化合物3的过程中,我们通过运用醋酸叔丁酯与70%HClO4的实验条件,一步反应实现了对L-赖氨酸羧基的Boc保护与α氨基的Boc脱保护。
如图3所示,I号路线为化合物3潜在的一般性合成路线,其合成过程为,首先对赖氨酸两个氨基进行保护,然后用叔丁基对羧基进行保护,最后选择性脱除α位Boc保护基得到化合物3。II号路线为笔者优化改进路线,两种路线相比,I号路线比II号路线多一个步骤,且由于叔丁醇的位阻较大,其与羧基的酯化反应将较为困难,在实际的合成过程中,其酯化产率低于30%,且后面还有一个Boc脱除的步骤,使化合物2到化合物3的产率低于20%。因此,我们设计了II号路线来达到合成化合物3的目的,首先仍然是对L-赖氨酸的两个氨基进行保护,在得到化合物2后,利用醋酸叔丁酯与70%HClO4与羧基进行酯交换反应得到L-赖氨酸叔丁酯衍生物。与此同时,在反应过程中,利用HClO4的强酸性,反应还伴随着氨基上的Boc脱除进而实现一步合成化合物3的目的,II号路线的设计不仅减少了反应步骤,而且将化合物3的产率从I号路线的小于20%提高到70%。

图 3 化合物3的合成优化策略Fig.3 Optimized strategy for the synthesis of compound 3
3 结论
本文提供了一种PSMA小分子抑制剂的合成策略,以期望用于辅助开发靶向前列腺癌的分子探针及靶向治疗药物。合成路线中所选用的原料便宜易得,反应过程条件温和,有着18%的反应总产率。同时,在开发PSMA小分子抑制剂的合成路线过程中,本文提供了一种一步法实现赖氨酸羧基与ε氨基Boc保护与脱保护的合成思路。
