RTM-GWAS方法应用于大豆RIL群体百粒重QTL检测的功效
2020-06-03潘丽媛贺建波赵晋铭王吴彬邢光南喻德跃张小燕李春燕陈受宜盖钧镒
潘丽媛,贺建波,赵晋铭,王吴彬,邢光南,喻德跃,张小燕,李春燕,陈受宜,盖钧镒
RTM-GWAS方法应用于大豆RIL群体百粒重QTL检测的功效
潘丽媛1,贺建波1,赵晋铭1,王吴彬1,邢光南1,喻德跃1,张小燕3,李春燕3,陈受宜2,盖钧镒1
(1南京农业大学大豆研究所/国家大豆改良中心/农业部大豆生物学与遗传育种重点实验室/作物遗传与种质创新国家重点实验室/江苏省现代作物 生产协同创新中心,南京 210095;2中国科学院遗传发育研究所/植物基因组学国家重点实验室,北京 100101;3山东圣丰种业科技有限公司,山东嘉祥 272400)
【】为全面解析大豆重组自交系群体中调控百粒重性状的QTL体系,将限制性两阶段多位点全基因组关联分析方法(RTM-GWAS)和不同定位方法进行比较、优选,为后续候选基因体系探索及分子标记辅助育种设计提供依据。利用以科丰1号和南农1138-2为亲本衍生的重组自交系群体NJRIKY的427个家系,通过由全基因组39 353个SNP构建的3 683个SNPLDB标记及3个环境下的百粒重表型数据,选用复合区间作图法(CIM)、基于混合线性模型的全基因组关联分析方法(MLM-GWAS)和RTM-GWAS 3种方法检测百粒重QTL,通过QTL数目和总的表型变异解释率比较检测功效,挑选最佳定位结果进行NJRIKY群体中的百粒重遗传体系解析。通过候选基因体系的功能注释,挖掘调控大豆百粒重的生物学途径。科丰1号与南农1138-2的百粒重差异较大,多环境平均数分别为9.0和17.9 g,遗传变异系数为12.4%,遗传率为85.4%,适用于百粒重性状的遗传解析。比较3种方法定位结果表明RTM-GWAS方法表现最佳,检测QTL数目最多(57个),解释表型变异最多(70.78%)。而CIM仅检测到14个QTL,解释了56.47%的表型变异,MLM-GWAS仅定位到6个QTL,解释了18.47%的表型变异。RTM-GWAS共检测到57个QTL,分布在19条染色体上,表型变异解释率为0.03%—7.57%,其中41个QTL覆盖了已报道的来自30个双亲群体的81个百粒重QTL,16个QTL为新发现位点,包含一个表型变异解释率大于3%的大效应位点。此外,检测的57个QTL中有20个位点与环境存在互作效应。这57个QTL构成了影响NJRIKY群体百粒重性状的遗传体系。通过SNPLDB标记与预测基因内的SNP进行χ2检验,共筛选到36个候选基因,其中4个候选基因来自大效应QTL,剩余32个候选基因来自小效应QTL。通过GO注释发现这些候选基因功能注释丰富,其中13个候选基因与籽粒发育直接相关,剩余的候选基因功能丰富,包含转运、转录调节因子等,表明不同生物学途径的基因共同调控NJRIKY群体中百粒重性状的表达。3种定位方法中,高效的RTM-GWAS方法检测到较为全面的NJRIKY群体的百粒重QTL,更适用于双亲RIL群体的QTL定位。不同功能的候选基因共同调控了复杂的百粒重性状的表达。
大豆;百粒重;QTL;重组自交家系;限制性两阶段多位点全基因组关联分析
0 引言
【研究意义】大豆是人类植物蛋白和脂肪的主要食物来源[1],产量一直是大豆生产发展的关注点,而百粒重是大豆产量构成的重要因子,一般与产量呈正相关[2]。百粒重作为数量性状,具有复杂的遗传基础,而现有的连锁定位方法,仅能定位到少数几个位点,限制了其遗传体系的全面解析,需要高效的定位方法进行全基因组QTL检测以全面解析其遗传基础。【前人研究进展】前人基于连锁定位的复合区间作图法(composite interval mapping,CIM)[3]对多个双亲群体进行了百粒重性状的QTL定位分析。目前,SoyBase数据库(https://www.soybase.org/,截至2017年3月)已经收录了基于40个不同群体、利用不同定位方法检测到的250个大豆百粒重QTL。最近,Fujii等[4]利用一个包含181个家系的重组自交系群体(recombinant inbred line,RIL),在2个环境下利用CIM方法共检测到5个百粒重QTL,分布于第12、13、17和20等4条染色体上。Zhang等[5]通过半野生豆花皮豆(Huapidou)和一个栽培豆齐黄26(Qihuang26)衍生的RIL群体,利用完备区间作图法(inclusive composite interval mapping,ICIM)和CIM分别检测到10个和5个百粒重QTL。由于连锁定位往往仅能定位到少数几个大效应位点,并且置信区间较大,不利于进一步筛选候选基因。现今,众多研究者通过关联分析的方法,利用自然群体进行大豆百粒重遗传解析[6-13]。Copley等[14]利用86个早熟组(MG 000—MG 00)大豆材料在2个环境下仅检测到5个百粒重QTL。Jing等[15]通过由地方种质和育成品种构成的包含185个大豆材料的自然群体检测到20个与百粒重关联的SNP位点,其中11个SNP连续分布在同一基因组区域。为了解决现有GWAS方法中遗传率缺失(missing heritability)和自然群体中的复等位变异问题,He等[16]开发了一种新的限制性两阶段多位点关联分析方法(restricted two-stage multi-locus genome- wide association study,RTM-GWAS),该方法通过遗传率控制位点的总表型变异解释率,利用二阶段方法和多位点模型提高了检测功效,并成功应用于多个自然群体和多亲本群体[17-22]。【本研究切入点】利用连锁定位方法检测到的大豆百粒重QTL较少,表型变异解释率丢失较多。而随着分子标记信息的加密,关联分析的精度提高,并且双亲群体中的群体结构简单[23],前人已多次成功将关联分析方法应用于双亲群体中[24-26],选择高效的关联分析方法有助于大豆百粒重数量性状遗传基础的全面解析,可为揭示其完整基因体系提供支持。【拟解决的关键问题】本研究基于科丰1号×南农1138-2衍生的重组自交系NJRIKY群体,利用3个环境下的大豆百粒重表型数据和3 683个SNPLDB标记,使用CIM、MLM-GWAS和RTM-GWAS 3种定位方法检测百粒重QTL,以QTL数目及总的表型变异解释率判定其检测功效,筛选最佳定位方法,进一步解析NJRIKY群体中百粒重的全基因组QTL体系及候选基因体系。
1 材料与方法
1.1 供试材料
以栽培大豆科丰1号和南农1138-2为母本和父本,衍生了一个含有427个家系的重组自交系(recombinant inbred line,RIL)群体。群体命名为NJRIKY,由两部分组成;其中184个家系创制于1994年夏,F2世代采用单粒传法繁代,F3-F7家系内混合收获播种,F7家系内随机收获1株,种植F2:8世代,最终经过群体校正后获得;另外243个家系采用同样方法获得[27]。亲本科丰1号来自黄淮海二熟制春夏作大豆品种生态区,由中科院遗传研究所选育,熟期组为MG II,黑色种皮;亲本南农1138-2来自长江中下游二熟制春夏作大豆品种生态区,由南京农业大学选育,熟期组为MG V,黄色种皮[28]。
1.2 田间试验
NJRIKY群体及亲本,2012—2013年夏季于江苏省南京市南京农业大学江浦试验站(32.07°N, 118.62°E)和2013年夏季于山东省济宁市圣丰试验站(36.67°N,116.98°E)3个环境进行田间试验。3个环境分别简称为12JP、13JP和13SF,代表2012年江浦试验站、2013年江浦试验站和2013年圣丰试验站。田间试验均采用立方格子设计,每个环境3次重复,江浦试验站采用穴播,两穴一个小区,穴距为0.7 m×0.8 m,每穴定苗6株;圣丰试验站采用行播,行长2 m,行距0.5 m,常规田间管理,籽粒成熟后按小区分别收获脱粒,并在35—40℃条件下烘干至恒重,取100个完整籽粒置于精度0.01 g天平称量记录。
1.3 标记分型
利用427家系及2个亲本的新鲜幼嫩叶片采用CTAB法[29]提取DNA,建库并利用Illumina Hiseq2000进行双端测序,通过与参考基因组Williams82 v1.1比对获得SNP。按照缺失≤20%、错误率≤1%和杂合率<5%的标准过滤获得39 353个高质量SNP。利用NPUTE[30]对SNP缺失基因型进行填补,用于SNPLDB(SNP linkage disequilibrium block)标记构建[25]。利用贺建波等[16]开发的RTM-GWAS软件,根据双亲带型划分SNPLDB标记,设置的最大窗口为200 kb使用默认设置的置信区间法定义区段,计算标记间的LD(D’),若95%以上的SNP的D’值位于0.70—0.98,则说明具有强LD水平,则被划分到一个单体型区块中,这样全基因组的SNP被划分为SNP连锁不平衡区块(SNP linkage disequilibrium block,SNPLDB)。根据双亲带型进行SNPLDB基因型分型,少量非亲本单体型被与其最相似的亲本单体型替换,最终获得3 683个SNPLDB标记用于遗传图谱构建及QTL定位[25]。
1.4 表型数据分析
采用SAS软件PROC GLM进行多环境下表型数据的联合方差分析,同时利用PROC MIXED的REML方法估计方差组分,根据和分别估计多环境和单环境下的性状遗传率,遗传变异系数(genotypic coefficient of variation,GCV)为100×σ/,其中为遗传方差,为基因型与环境互作方差,为误差方差,为环境的个数,为一个环境内的试验重复数,为整体平均数。
1.5 QTL定位方法
利用3种方法进行NJRIKY群体的百粒重QTL定位,分别为复合区间作图法(CIM)[3]、混合线性模型全基因组关联分析方法(mixed linear model GWAS或MLM-GWAS)[31]、限制性两阶段多位点全基因组关联分析方法(RTM-GWAS)[16]。
CIM方法应用Windows QTL Cartographer V2.5软件,表型数据为多环境下各家系百粒重均值,前景及背景选择的显著水平为0.05,步长为1 cM,并进行排列测验1 000次,确定QTL检测LOD阈值。
MLM-GWAS方法[31]应用TASSEL 3.0软件[32],表型数据为多环境下各家系百粒重均值,参数设定为默认参数,采用5个主成分(principal component,P)和个体间亲缘关系(kinship,K)矩阵,即P+K模型的MLM进行关联分析,并利用R软件fdrtool包[33]进行假发现率(false discovery rate,FDR)多重测验校正,显著水平为0.05。
RTM-GWAS方法利用限制性二阶段多位点全基因组关联分析方法(RTM-GWAS)软件[16]进行QTL定位,表型数据为多环境小区原始观测值,显著水平设为0.05,分析模型同时考虑QTL与环境互作效应。根据表型变异解释率(phenotypic variation explained,PVE)大小将QTL分为两类:大贡献(large contribution,LC)主效位点(PVE≥3%);小贡献(small contribution,SC)主效位点(PVE<3%)。
1.6 候选基因体系注释
首先,检测SNPLDB标记(±100 kb)物理区间内的注释基因;然后利用卡平方(χ2)独立性测验,显著水平设为0.05,检测SNPLDB标记与注释基因内SNP之间的相关性。当这二者显著相关时,则该SNP所在的注释基因被确定为候选基因;同时,利用SoyBase数据库(https://www. soybase.org)提取所有注释基因的功能注释(gene ontology,GO),优先选择与籽粒发育相关的注释基因确定为候选基因。同时给出候选基因的功能注释(GO)结果。
2 结果
2.1 多环境下NJRIKY群体百粒重变异
从表1可以看出3个环境下NJRIKY群体的亲本科丰1号和南农1138-2的表型差异较大,多环境平均值分别为9.0和17.9 g,重组自交家系的百粒重变异范围为8.6—17.1 g(图1),遗传变异系数为12.4%,变异范围位于双亲之间。该群体百粒重的遗传率较高,多环境联合下为85.4%,单环境下为92.5%—93.9%。多环境联合方差分析的结果表明家系间、环境间及家系与环境互作的差异均达到极显著水平(表2)。

Mean表示各家系3个环境百粒重平均数
表1 多环境下NJRIKY群体百粒重的次数分布和描述统计
Table 1 Frequency distribution and descriptive statistics of 100-seed weight of NJRIKY under multiple environments

:遗传变异系数: genotypic coefficient of variation
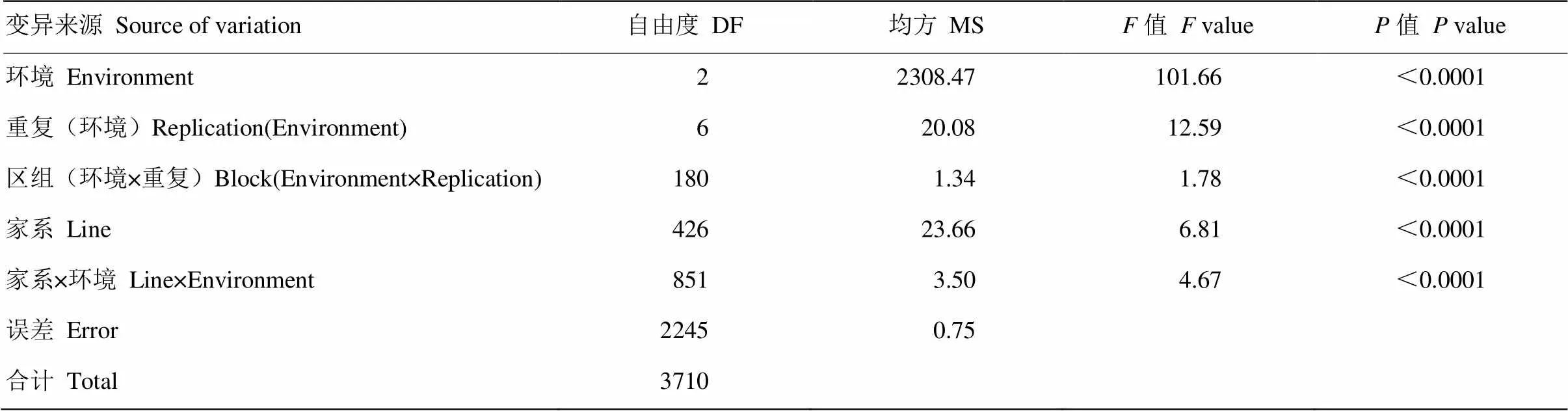
表2 NJRIKY群体多环境联合方差分析结果
2.2 不同定位方法下百粒重QTL检测结果
利用3 683个SNPLDB标记及CIM、MLM-GWAS和RTM-GWAS 3种定位方法对NJRIKY群体的百粒重性状进行QTL定位(表3),并通过QTL的数目和总的表型变异解释率来比较各个方法的检测功效,选取最佳的定位结果进行该群体的百粒重遗传体系解析。

表3 NJRIKY群体中利用不同定位方法检测到的百粒重位点结果概要
检测的QTL:本研究检测的QTL数目和染色体数目,57(19):57个QTL位于19条染色体上。Mapped QTL:所有定位到的QTL的汇总。大贡献主效QTL和小贡献主效QTL分别表示贡献率≥3%与<3%的主效QTL。QTL×Env.表示QTL与环境互作效应。已报道的QTL:SoyBase (http://www.soybase.org) 数据库中前人报道的与始花期位于邻近1 Mb位置的QTL数目,41(84)表示41个QTL与SoyBase (http://www.soybase.org) 中84个QTL位置一致或邻近
Detected QTLs: the number of detected QTLs and chromosomes in the present study, 57(19): 57 QTLs on 19 chromosomes. Mapped QTL: the sum of all major QTLs. LC major QTL: the QTL detected from RTM-GWAS with its PVE more than 3% is called large-contribution (LC) major QTLs; SC major QTL: small-contribution major QTL with PVE less than 3%. QTL × Env.: the interaction between QTL and environments. Reported QTLs: the number of QTLs reported in SoyBase (http://www.soybase.org) that is nearby the present detected QTL in the RTM-GWAS procedure according to the physical position within 1 Mb, 41 (84): 41 QTLs shared same confidence regions with 84 SoyBase QTLs
在连锁定位中较为普遍使用的CIM方法下共检测到14个百粒重QTL,分布在8个连锁群上,总表型变异解释率(PVE)为56.47%(图2和表4)。在普遍使用的关联分析方法MLM-GWAS中仅检测到6个QTL,位于3条染色体上,总PVE为18.47%(图2和表4)。利用RTM-GWAS方法,检测到57个百粒重QTL,分布在除Gm19以外的19条染色体上,共解释70.78%的表型变异,结合方差分析估计得到的遗传率为85.4%,剩余14.52%的表型变异来源于未定位到的微效QTL(图2)。其中位于13条染色体上的20个百粒重QTL与环境存在互作效应,共解释4.20%的表型变异(表3和表5)。
遵循常规,以位点前后1 Mb的范围来归并QTL,发现RTM-GWAS方法几乎覆盖了CIM(8个QTL/共14个QTL,PVE=40.85%/总PVE=56.47%)和MLM-GWAS(4个QTL/共6个QTL,PVE=12.96%/总PVE=18.47%)方法的结果,包括位于第11染色体上的效应最大的位点Gm11_BLOCK_5228756_ 5269867,这个位点在3种方法中的表型变异解释率均最大,分别为11.05%(CIM)、3.69%(MLM-GWAS)和7.57%(RTM-GWAS)。

表4 NJRIKY群体中在CIM和MLM-GWAS方法下的百粒重QTL
LOD/-lg:LOD为CIM方法中QTL检测的LOD(logarithm of odds)得分,-lg为MLM-GWAS方法中QTL检测的值的对数值。SoyBase QTL:SoyBase (http://soybase.org) 数据库中前人报道的与始花期位于邻近1 Mb位置的QTL名称
LOD/-lg: LOD is the logarithm of odds score in CIM and -lgis thevalue in log10 scale in MLM-GWAS. SoyBase QTL: the QTL names reported in SoyBase (http://soybase.org) that is nearby the present detected QTL in the RTM-GWAS procedure according to the physical position within 1 Mb

表5 NJRIKY群体中与百粒重性状关联的QTL位点
*表示该QTL具有QTL与环境互作效应。标记中Gm后的数字表示其所在染色体,末端数字为其对应物理位置
* indicates the QTL interacted with environments. The number after “Gm” means the chromosome number, the number at the end of a marker indicates the corresponding physical position
以上3种定位方法的百粒重QTL结果表明,RTM-GWAS方法的检测功效最高,检测到更多的QTL(57 vs. 14和6),解释了更多的表型变异(70.78% vs. 56.47%和18.47%),在NJRIKY群体中检测到了较为全面的百粒重性状的遗传体系,所以将对RTM-GWAS方法检测到的由57个QTL构成的影响NJRIKY群体百粒重性状的遗传体系进行进一步的遗传解析。这里要特别指出,RTM-GWAS还检测到57个QTL中有20个是与环境存在互作(或因环境而波动)的QTL,这是其他2种定位方法所不及的。
2.3 NJRIKY RIL群体中百粒重的QTL及候选基因体系
利用3 683个SNPLDB标记及RTM-GWAS方法对NJRIKY群体百粒重进行全基因QTL检测,第一阶段中,利用一般线性模型(general linear model,GLM)筛选到1 860个候选SNPLDB标记;第二阶段中,多位点模型最终检测到57个与百粒重相关的QTL(图3和表5)。

a:染色体结构,异染色质区域标为浅红色(单位为Mb)

图3 NJRIKY中百粒重关联分析的曼哈顿图和QQ图
57个百粒重QTL分布于19条染色体,表型变异解释率变化范围为7.57%—0.13%,其中35个QTL的PVE为0.13%—1.00%,17个为1.00%—3.00%,剩余5个为3.00%—7.57%,表明百粒重作为一个复杂的数量性状,其所包含的位点贡献率变化范围广,不仅仅包含表型变异解释率很大的QTL,还包含更多小贡献率的QTL。
57个QTL构成的调控NJRIKY群体百粒重的QTL体系中,第4染色体包含的QTL数目最多,共14个QTL,第20染色体包含的QTL最少,仅为1个QTL。、、、、分别解释了7.57%、5.12%、3.72%和3.61%的表型变异(phenotypic variation,PV),为大贡献主效QTL,共解释了23.30%的PV,剩余52个小贡献主效QTL共解释了47.48%的PV。NJRIKY群体的QTL体系中41个QTL与SoyBase中收录的包含52个亲本的30个双亲群体中检测到的81个QTL(共250个QTL)的位置一致(表3)。
RTM-GWAS在3个环境下还检测到20个QTL与环境存在互作效应,其中,3个位于第4、9和11染色体的大贡献主效QTL和17个位于12条染色体上的小贡献主效QTL(表5)。
通过RTM-GWAS方法对NJRIKY群体的百粒重性状进行较为全面的检测后,基于高密度的遗传标记,进一步推断其候选基因体系。通过SNPLDB标记与预测基因内的SNP进行χ2检验,共在57个QTL中检测到36个候选基因解释了54.12%的PV,其中4个候选基因来自4个大贡献主效QTL,剩余32个候选基因来自32个小贡献主效位点,还有20个QTL中未检测到符合条件的候选基因(存在2个QTL的候选基因相同)(表6)。通过GO注释可以看出13个候选基因与籽粒发育相关,剩余的候选基因的功能变化丰富,包含了转运、蛋白糖基化、转录调节因子、脱落酸响应等,表明这些候选基因通过不同生物途径共同调控百粒重性状的表达。
3 讨论
3.1 高效的RTM-GWAS方法适用于双亲群体的QTL定位
随着测序技术的快速发展,植物生物学家的研究重心逐渐从少数基因功能向挖掘调控目标性状的全面基因体系和基因网络转移[34]。前人结果表明,RTM-GWAS方法适用于自然群体和巢式关联群体的数量性状全基因组位点检测[16-17, 19]。本研究在3种不同的QTL方法中,RTM-GWAS几乎覆盖了其他方法(CIM和MLM-GWAS)检测到的所有QTL。RTM-GWAS检测到较多的QTL(57个QTL)解释了更多的表型变异(70.78%),而在CIM和MLM-GWAS检测到的QTL仅解释了56.47%和18.47%的表型变异。由于双亲群体中不存在复杂的群体结构问题,将关联分析的方法应用到双亲群体中,提高了检测精度,将大大加快双亲群体数量性状的遗传体系的全面解析。另外,利用RTM-GWAS方法,由于其通过遗传率控制了位点的总表型变异解释率,利用二阶段和多位点模型提高了检测功效,仅利用一个双亲群体就检测到了41个QTL与已报道的52个大豆材料中的81个QTL位置一致,并且13个候选基因与籽粒发育相关,表明RTM-GWAS方法在双亲群体RIL群体中QTL定位表现高效,适用于双亲群体的QTL定位。
3.2 大豆百粒重性状的遗传体系
大豆是人类重要的植物蛋白和油脂的食物来源,大豆百粒重与产量的直接相关[2],直接决定了大豆百粒重性状的重要性。前人已经通过不同类型的群体对大豆百粒重的遗传体系进行了研究[18, 35],本研究利用RTM-GWAS方法在亲本具有较大差异的NJRIKY群体中检测到57个百粒重相关联的QTL,其中16个QTL是该群体检测到的新位点,的PVE达到5.12%。5个大效应主效QTL中,的贡献率最高为7.57%,其位置与SoyBase中的、和一致,其中和来源于科丰1号和南农1138-2的184个家系后代群体[36-37],QTL区间较大(5 132 968—6 184 217 bp),群体扩大及标记加密后,SNPLDB的位置为5 228 756-5 269 867 bp。的贡献率为3.71%,与已报道的和位于相近位置。的PVE为3.60%,与已报道的3个QTL位置相近,其中的定位群体为NJRIKY群体的184个家系。的贡献率为3.27%,与和[38-39]的位置相近,这两个已报道位点的标记区间极大,约7 Mb,可能是定位的群体大小及标记密度的影响。其他小贡献主效QTL中也不乏被多个群体多次定位到的位点,如的贡献率在本群体中仅为0.68%,其与SoyBase中的7个双亲群体中的8个QTL位置相近。

表6 RTM-GWAS方法中检测到的与百粒重相关的候选基因体系
加粗的候选基因表示该基因的GO注释与籽粒发育相关
The candidate genes in boldface mean their gene ontology description is associated with seed development
NJRIKY群体中检测到36个候选基因中,13个候选基因与籽粒发育相关。其中2个候选基因、分别来自2个大贡献主效QTL——和,剩余11个候选基因来自11个小贡献主效位点。如,(PVE=1.87%)的候选基因与胚发育相关,其在拟南芥中的同源基因表达产物为TOR蛋白,直接调控了植物的胚生长发育和逆境响应[40]。具有不同功能的候选基因,共同调控了复杂的百粒重性状的表达,而现今的研究还具有局限性,包括材料的限制等,因此,综合越来越多的群体中百粒重性状的结果,结合高效的QTL定位方法,为全面解析基因体系及网络提供了必要基础。
4 结论
NJRIKY RIL群体中,双亲的百粒重差异较大,适用于百粒重性状的遗传解析。利用3种定位方法,在3个环境下,RTM-GWAS方法定位到的QTL更多,表型变异解释率更高,其检测功效优于其他2种方法(CIM和MLM-GWAS)。通过RTM-GWAS定位到57个百粒重QTL构成了NJRIKY群体的百粒重遗传体系,表明高效的QTL定位方法将加快全面解析复杂的百粒重性状的遗传体系。
[1] SMITH T J, CAMPER H M. Effect of seed size on soybean performance., 1970, 67(5): 681-684.
[2] BURRIS J S, EDJE O T, WAHAB A H. Effects of seed size on seedling performance in soybeans: II. seedling growth and photosynthesis and field performance1., 1973, 13(2): 207.
[3] ZENG Z. Precision mapping of quantitative trait loci., 1994, 136(4): 1457-1468.
[4] FUJII K, SAYAMA T, TAKAGI K, TAKAGI K, KOSUGE K, OKANO K, KAGA A, ISHINOTO M. Identification and dissection of single seed weight QTLs by analysis of seed yield components in soybean., 2018, 68(2): 177-187.
[5] ZHANG Y, LI W, LIN Y, ZHANG L, WANG C, XU R. Construction of a high-density genetic map and mapping of QTLs for soybean () agronomic and seed quality traits by specific length amplified fragment sequencing., 2018, 19(1): 641.
[6] ZHOU Z, JIANG Y, WANG Z, GOU Z, LYU J, LI W, YU Y, SHU L, ZHAO Y, MA Y, FANG C, SHEN Y, LIU T, LI C, LI Q, WU M, WANG M, WU Y, DONG Y, WAN W, WANG X, DING Z, GAO Y, XIANG H, ZHU B, LEE S H, WANG W, TIAN Z. Resequencing 302 wild and cultivated accessions identifies genes related to domestication and improvement in soybean., 2015, 33(4): 408-414.
[7] ZHANG H, HAO D, SITOE H M, YIN Z, HU Z, ZHANG G, YU D, SINGH R. Genetic dissection of the relationship between plant architecture and yield component traits in soybean () by association analysis across multiple environments., 2015, 134(5): 564-572.
[8] CONTRERAS-SOTO R I, MORA F, DE OLIVEIRA M A, HIGASHI, W, SCAPIM C A, SCHUSTER I A. genome-wide association study for agronomic traits in soybean using SNP markers and SNP-based haplotype analysis., 2017, 12(2): e0171105.
[9] HU Z, ZHANG D, ZHANG G, KAN G, HONG D, YU D. Association mapping of yield-related traits and SSR markers in wild soybean (Sieb. and Zucc.)., 2014, 63(5): 441-449.
[10] ZHANG J, SONG Q, CREGAN P B, JIANG G. Genome-wide association study, genomic prediction and marker-assisted selection for seed weight in soybean ()., 2016, 129(1): 117-130.
[11] SONAH H, O'DONOUGHUE L, COBER E, RAJCAN I, BELZILE F. Identification of loci governing eight agronomic traits using a GBS-GWAS approach and validation by QTL mapping in soya bean., 2015, 13(2): 211-221.
[12] YAN L, HOFMANN N, LI S, FERREIRA M E, SONG B, JIANG G, REN S, QUIGLEY C, FICKUS E, GREGAN P, SONG Q. Identification of QTL with large effect on seed weight in a selective population of soybean with genome-wide association and fixation index analyses., 2017, 18(1): 529.
[13] WANG J, CHU S, ZHANG H, ZHU Y, CHENG HAO, YU D. Development and application of a novel genome-wide SNP array reveals domestication history in soybean., 2016, 6: 20728.
[14] COPLEY T R, DUCEPPE M O, O'DONOUGHUE L S. Identification of novel loci associated with maturity and yield traits in early maturity soybean plant introduction lines., 2018, 19(1): 167.
[15] JING Y, ZHAO X, WANG J, TENG W, QIU L, HAN Y, LI W. Identification of the genomic region underlying seed weight per plant in soybean (L. Merr.) via high-throughput single- nucleotide polymorphisms and a genome-wide association study., 2018, 9: 1392.
[16] HE J, MENG S, ZHAO T, XING G, YANG S, LI Y, GUANG R, LU J, WANG Y, XIA Q, YANG B, GAI J. An innovative procedure of genome-wide association analysis fits studies on germplasm population and plant breeding., 2017, 130(11): 2327-2343.
[17] LI S, CAO Y, HE J, ZHAO T, GAI J. Detecting the QTL-allele system conferring flowering date in a nested association mapping population of soybean using a novel procedure., 2017, 130(11): 2297-2314.
[18] ZHANG Y, HE J, WANG Y, XING G, ZHAO J, LI Y, YANG S, PALMER R G, ZHAO T, GAI J. Establishment of a 100-seed weight quantitative trait locus-allele matrix of the germplasm population for optimal recombination design in soybean breeding programmes., 2015, 66(20): 6311-6325.
[19] MENG S, HE J, ZHAO T, XING G, LI Y, YANG S, LU J, WANG Y, GAI J. Detecting the QTL-allele system of seed isoflavone content in Chinese soybean landrace population for optimal cross design and gene system exploration., 2016, 129(8): 1557-1576.
[20] ZHANG Y, HE J, WANG H, WANG H, MENG S, XING G, LI Y, YANG S, ZHAO J, ZHAO T, GAI J. Detecting the QTL-allele system of seed oil traits using multi-locus genome-wide association analysis for population characterization and optimal cross prediction in soybean., 2018, 9: 1793.
[21] ZHANG Y, HE J, MENG S, LIU M, XING G, LI Y, YANG S, YANG J, ZHAO T, GAI J. Identifying QTL-allele system of seed protein content in Chinese soybean landraces for population differentiation studies and optimal cross predictions., 2018, 214(9),157.
[22] KHAN M A, TONG F, WANG W, HE J, ZHAO T, GAI J. Analysis of QTL-allele system conferring drought tolerance at seedling stage in a nested association mapping population of soybean [(L.) Merr.] using a novel GWAS procedure., 2018, 248(4): 947-962.
[23] MACKAY I, POWELL W. Methods for linkage disequilibrium mapping in crops., 2007, 12(2): 57-63.
[24] MALOSETTI M, VAN EEUWIJK F A, BOER M P, CASAS A M, ELIA M, MORALEJO M, BHAT P R, RAMSAY L, MOLINA CONO J L. Gene and QTL detection in a three-way barley cross under selection by a mixed model with kinship information using SNPs., 2011, 122(8): 1605-1616.
[25] PAN L, HE J, ZHAO T, XING G, WANG Y, YU D, CHEN S, GAI J. Efficient QTL detection of flowering date in a soybean RIL population using the novel restricted two-stage multi-locus GWAS procedure., 2018, 131(12): 2581-2599.
[26] RIYAN C, LIM J E, SAMOCHA K E, SOKOLOFF G, ABNEY M, SKOL A D, PALMER A A. Genome-wide association studies and the problem of relatedness among advanced intercross lines and other highly recombinant populations., 2010, 185(3): 1033.
[27] 王永军, 喻德跃, 章元明, 陈受宜, 盖钧镒. 重组自交系群体的检测调整方法及其在大豆NJRIKY群体的应用. 作物学报, 2004, 30(5): 413-418.
WANY Y J, YU D Y, ZHANG Y M, CHEN S Y, GAI J Y. Method of evaluation and adjustment of recombinant inbred line population and its application to the soybean RIL population NJRIKY.,2004, 30(5): 413-418. (in Chinese)
[28] 盖钧镒, 邱家驯, 赵团结. 大豆品种南农493-1和南农1138-2与其衍生新品种的亲缘关系及其育种价值分析. 南京农业大学学报, 1997, 20 (1): 1-8.
GAI J Y, QIU J X, ZHAO T J. An analysis of genetic relationship of nannong493-1 and nannong 1138-2 with their derivative cultivars and their potential in future breeding., 1997, 20(1): 1-8. (in Chinese)
[29] MURRAY M G, THOMPSON W F. Rapid isolation of high molecular weight plant DNA., 1980, 8(19): 4321-4325.
[30] ROBERTS A, MCMILLAN L, WANG W, PARKER J, RUSYN I, THREADGILL D. Inferring missing genotypes in large SNP panels using fast nearest-neighbor searches over sliding windows., 2007, 23(13): 401-407.
[31] YU J M, PRESSOIR G, BRIGGS W H, BI I V, YAMASAKI M, DOEBLEY J F, MCMULLEN M D, GAUT B S, NIELSEN D M, HOLLAND J B, KRESOVICH S, BUCKLER E S. A unified mixed-model method for association mapping that accounts for multiple levels of relatedness., 2006, 38(2): 203-208.
[32] BRADBURY P J, ZHANG Z, KROON D E, CASSTEVENS T M, RAMDOSS V, BUCKER E S. TASSEL: Software for association mapping of complex traits in diverse samples., 2007, 23(19): 2633-2635.
[33] STRIMMER K. fdrtool: A versatile R package for estimating local and tail area-based false discovery rates., 2008, 24(12): 1461-1462.
[34] GAI J. Quantitative Inheritance//MALOY S, HUGHES K. eds.. 2nd ed. San Diego: Academic Press, 2013: 18-21.
[35] LU X, XIONG Q, CHENG T, LI Q, LIU X, BI Y, LI W, ZHANG W, MA B, LAI Y, DU W, MAN W, CHEN S, ZHANG J. A PP2C-1 allele underlying a quantitative trait locus enhances soybean 100-seed weight., 2017, 10(5): 670-684.
[36] GAI J, WANG Y, WU X, CHEN S. A comparative study on segregation analysis and QTL mapping of quantitative traits in plants-with a case in soybean., 2007, 1(1): 1-7.
[37] ZHANG W K, WANG Y J, LUO G Z, ZHANG J, HE C, WU X, GAI J, CHEN S. QTL mapping of ten agronomic traits on the soybean (L. Merr.) genetic map and their association with EST markers., 2004, 108(6): 1131-1139.
[38] HAN Y, LI D, ZHU D, LI H, LI X, TENG W, LI W. QTL analysis of soybean seed weight across multi-genetic backgrounds and environments., 2012, 125(4): 671-683.
[39] CHEN Q, ZHANG Z, LIU C, XIN D, QIU H, SHAN D, SHAN C, HU G. QTL analysis of major agronomic traits in soybean., 2007, 6(4): 399-405.
[40] WANG P, ZHAO Y, LI Z, HSU C, LIU C, FU L, HOU Y, DU Y, XIE S, ZHANG C, GAO J, CAO M, HUANG X, ZHU Y, TANG K, WANG X, TAO W, XIONG Y, ZHU J. Reciprocal regulation of the TOR kinase and ABA receptor balances plant growth and stress response., 2018, 69(1): 100-112.
Detection power of RTM-GWAS applied to 100-seed weight QTL identification in a recombinant inbred lines population of soybean
PAN LiYuan1, HE JianBo1, ZHAO JinMing1, WANG WuBin1, XING GuangNan1, YU DeYue1, ZHANG XiaoYan3, LI ChunYan3, CHEN ShouYi2, GAI JunYi1
(1Soybean Research Institute, Nanjing Agricultural University /National Center for Soybean Improvement /Key Laboratory of Biology and Genetic Improvement of Soybean (General), Ministry of Agriculture/State Key Laboratory for Crop Genetics and Germplasm Enhancement/Jiangsu Collaborative Innovation Center for Modern Crop Production, Nanjing 210095;2Institute of Genetics and Developmental Biology, Chinese Academy of Sciences/State Key Laboratory of Plant Genomics, Beijing 100101;3Shandong Shofine Seed Technology Co. Ltd., Jiaxiang 272400, Shandong)
【】To thoroughly dissect the QTL system conferring 100-seed weight in a recombinant inbred lines population, the restricted two-stage multi-locus genome-wide association analysis (RTM-GWAS) method was compared with other mapping methods for method optimization, which will provides basis for further exploration of candidate gene system and molecular marker-assisted design breeding. 【】 A recombinant inbred line population consisting of 427 lines derived from a cross between Kefeng-1 and NN1138-2 was tested for its 100-seed weight under three environments. A total of 3 683 SNPLDBs (SNP linkage disequilibrium blocks) composed of 39 353 SNPs were applied to QTL mapping using three different mapping procedures, including the composite interval mapping (CIM) method, the mixed linear model (MLM-GWAS) method and the RTM-GWAS method, and the best mapping procedure was selected for the analysis of the 100-seed weight genetic system in NJRIKY population through comparing their detection power, including the detected number of QTLs and total phenotypic variation explained. 【】The 100-seed weight of Kefeng-1 and NN1138-2 were 9.0 g and 17.9 g, respectively, showing significant difference. The genotypic coefficient of variation and heritability of the trait were 12.4% and 85.4%, respectively. These results indicated that the population was suitable for genetic analysis of 100-seed weight trait. The RTM-GWAS procedure performed the best with the largest number of QTLs (57) explaining the most phenotypic variation (PVE=70.78%), while a total of 14 and 6 QTLs contributing 56.47% and 18.47% phenotypic variation were detected using CIM and MLM-GWAS, respectively. The 57 QTLs detected from the RTM-GWAS distributed on 19 chromosomes, of which 41 QTLs overlapped with 81 QTLs identified from 30 bi-parental populations in the literature. Furthermore, the PVE of 57 QTLs ranged from 0.03% to 7.57%, of which 16 QTLs were novel ones, including one large contribution major QTL(PVE>3%). Furthermore, 20 QTLs had significant interaction effect with environment. A total of 36 candidate genes were annotated from 37 QTLs through χ2test between SNPLDB markers and SNPs harboring in the predicted genes, of which 4 candidate genes were from the large contribution QTLs and other 32 candidate genes were from the small contribution QTLs. These candidate genes were included in different biological processes, of which 13 candidate genes were grouped in seed development directly, and the remaining candidate genes were grouped into different functions, such as transport, transcriptional regulators, etc., indicating that these genes from different biological pathways regulate the expression of 100-seed weight trait in NJRIKY together. 【】Among the three different mapping procedures, RTM-GWAS procedure is the most powerful one which can provide a relatively thorough detection of 100-seed weight QTLs in NJRIKY population, therefore, it is more suitable for QTL mapping in bi-parental population such as RIL population. The candidate genes with various functions jointly regulated the complex expression of 100-seed weight trait.
soybean [(L.) Merr.]; 100-seed weight; QTL (quantitative trait locus); recombinant inbred lines population; restricted two-stage multi-locus genome-wide association analysis

10.3864/j.issn.0578-1752.2020.09.004
2019-08-24;
2020-01-02
国家自然科学基金(31701447)、国家作物育种重点研发计划(2017YFD0101500,2017YFD0102002)、长江学者和创新团队发展计划(PCSIRT_17R55)、教育部111项目(B08025)、中央高校基本科研业务费项目(KYT201801)、农业部国家大豆产业技术体系CARS-04、江苏省优势学科建设工程专项、江苏省JCIC-MCP项目
潘丽媛,E-mail:panly89@126.com。通信作者贺建波,E-mail:hjbxyz@gmail.com。通信作者盖钧镒,E-mail:sri@njau.edu.cn
(责任编辑 李莉)
