糖尿病大鼠心肌损伤中程序性坏死通路相关蛋白的表达
2020-06-02王洪巨康品方
孙 硕,戎 李,莫 辰,张 冰,高 琴,王洪巨,康品方,3,张 恒
RIPK1-RIPK3-MLKL通路是近年来热度较高的细胞程序性坏死(Necroptosis)的经典通路。Necroptosis兼具有程序性调控以及细胞坏死的病理学特征[1-2],且依赖于RIPK3和p-MLKL的活性,这有别于既往我们所熟知的细胞死亡方式,如坏死、凋亡、自噬等。Necroptosis是一种重要的致死性信号通路,参与了各种疾病的发展,包括缺血性损伤、病毒感染、肝脏疾病、肿瘤等[3]。在应激条件下,RIPK3可以迁移到内质网上,增加活性氧(ROS)的生成,最终导致心肌细胞因能量不足而膨胀和破裂[4]。糖尿病心肌病(DCM)是指有糖尿病基础,在心脏微血管病变及代谢紊乱基础上引发心肌广泛缺血坏死,其主要病理过程涉及心肌细胞膜钙通道转运、心肌间质纤维化、冠状动脉微血管病变、RAS系统激活等多方面[5]。有研究表明PKCβ/p66氧化应激途径[6]、caspase-1炎症通路[7]等与糖尿病心肌损伤关系密切。RIPK3-CaMKII通路也被证实参与心肌损伤过程[8]。因此为了更好地理解Necroptosis在DCM发生发展中的作用和调节机制,本实验通过复制糖尿病模型,观测Necroptosis通路中主要因子及丙二醛的含量变化,观察糖尿病心肌损伤中Necroptosis如何变化及其可能的调控机制。
1 材料与方法
1.1 实验材料及模型复制 选取SD大鼠24只,体质量(BW)210~245 g,平均BW(229.78±22.41)g,均来自蚌埠医学院实验动物中心同一批次大鼠,每笼4~5只,饮食自由,室温恒定,昼夜规律,空气湿度为40%~60%。适应性喂养1周后,随机选取6只为CON组。其余18只予以高脂高糖饮食(HC组+DM组),饲料配方为:64.75%基础饲料+10%猪油+20%白砂糖+5%蛋白粉+0.05%维生素+0.2%矿物质。3周后随机选取12只大鼠为DM组,禁食12 h后腹腔注射链脲佐菌素(STZ)40 mg/kg造模,72 h后检测空腹血糖,连续3 d,空腹血糖≥16.7 mmol/L,尿糖3+~4+,说明造模成功。每组留取6只,其余备用。此后每3 d检测尿糖和血糖,剔除不达标大鼠。死亡大鼠由同批次大鼠代替。10周后统一处理大鼠。主要试剂:STZ(美国Sigma公司),兔抗大鼠RIP1、RIP3、MLKL抗体(美国Abcam公司),MDA试剂盒(南京建成公司),DMSO(美国Sigma公司),Trizol Reagent(美国Invitrogen公司),SDS-PAGE缓冲液(上海碧云天生物技术有限公司),高速离心机(上海卢湘离心机仪器有限公司)。
1.2 计算大鼠心体比 大鼠称BW后采用4%水合氯醛按照8 mL/kg进行腹腔注射,麻醉满意后,仰卧固定于手术台上,沿前正中线切开,打开心腔,剥离心脏,0.9%氯化钠溶液冲净血液,剪除周围纤维组织及大血管,滤纸吸干后天平称全心质量(HW),心体比为HW/BW。
1.3 测量大鼠心功能 大鼠处死前统一麻醉后行小动物超声,在左心长轴切面上测量左心室舒张末期内径(LVEDD)、左心室收缩末期内径(LVESD);通过M型超声获得左心室射血分数(LVEF),每只测量3次取平均值,为减少大鼠个体差异所造成的误差,采用体表面积进行校正,体表面积(m2)=0.09×[BW(kg)]2/3。
1.4 ELISA检测肿瘤坏死因子(TNF-α)、白细胞介素(IL-6)及丙二醇(MDA)含量 打开心腔后,从腹主动脉抽取1~2 mL血液,4 ℃ 2 000 r/min离心10 min后取血浆,-80 ℃保存。用时水浴溶解,按照试剂盒说明书,加样、洗板、封闭、加检测抗体、洗板、加酶、洗板、显色、终止反应、读板、获取标准曲线。
1.5 RT-PCR检测RIPK1、RIPK3、MLKL的RNA含量 心脏称重后取0.1 g×2份心尖部心肌组织,其中1份裂解匀浆,4 ℃ 10 000 r/min 5 min后取上清液分别分成2份至新的EP管中。TRIzol法提取总RNA,酶标仪Take3测RNA纯度及浓度;按照说明书,冰上进行RNA逆转录,得到逆转录产物cDNA;溶解引物后分装,引物的信息见表1;加样,设计反应程序,以CON组作为对照,计算得到2-ΔΔCt值,进行数据分析。

表1 RIPK1、RIPK3、MLKL的上、下游引物序列表
1.6 蛋白印迹检测RIPK1、RIPK3、MLKL的蛋白含量 取每组心肌细胞上清液1份,BCA法蛋白定量后于-20 ℃保存;凝胶制备,SDS PAGE电泳;并将胶用半干转膜法转移至激活后的 PVDF膜;配置封闭液,封闭,加入稀释的一抗,孵育,加入稀释的二抗,孵育,TBST洗膜;化学发光剂显色1 min,BIO-RAD凝胶成像系统曝光并获取图像;经凝胶成像系统测量条带灰度值,计算实验蛋白的相对表达量。
1.7 心肌Masson染色 取备用心尖组织1份,4%多聚甲醛立即固定,包埋切片,行Masson 染色。Masson染色主要的步骤为:石蜡切片脱蜡至水;自来水和蒸馏水反复冲洗;Regaud苏木精染液染核5~10 min;充分水洗;用Masson丽春红酸性复红液5~10 min;以2%冰醋酸水溶液浸洗15 min;1%磷钼酸水溶液分化3~5 min;苯胺蓝或光绿液染5 min;0.2%冰醋酸水溶液浸洗15 min。95%乙醇、无水乙醇、二甲苯透明、中性树胶封固。
1.8 统计学方法 采用方差分析和q检验。
2 结果
2.1 一般情况 CON组及HC组均未出现大鼠死亡,CON组大鼠毛色光泽,行动敏捷,较为活泼,HC组大鼠外形略显肥胖。DM组死亡4只,出现明显的“三多一少”症状,外形瘦小,蜷卧时间增加,尿中骚味明显,更换敷料频率增加。
2.2 大鼠HW/BW比较 与CON组比较,HC组HW明显增加(P<0.01),BW和HW/BW无明显变化(P>0.05);与HC组比较,DM组HW减轻,BW下降,HW/BW明显增加(P<0.05~P<0.01)(见表2)。
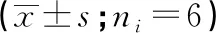
表2 大鼠HW、BW、HW/BW比较
q检验:与CON组比较**P<0.01;与HC组比较#P<0.05,##P<0.01
2.3 大鼠LVEDD、LVESD及LVEF变化 与CON组比较,HC组的LVESD、LVEF无明显差异(P>0.05),未见明显心功能不全;DM组较CON组及HC组,LVEDD及 LVESD均扩大,LVEF降低(P<0.01),心功能明显下降(见图1、表3)。
表3 大鼠LVEDD、LVESD及LVEF比较
q检验:与CON组比较**P<0.01;与HC组比较##P<0.01
2.4 血浆TNF-α、IL-6及MDA水平 HC组TNF-α、IL-6及MDA水平均高于CON组(P<0.05~0.01);DM组的TNF-α、IL-6及MDA水平均高于CON组和HC组(P<0.01)(见表4)。
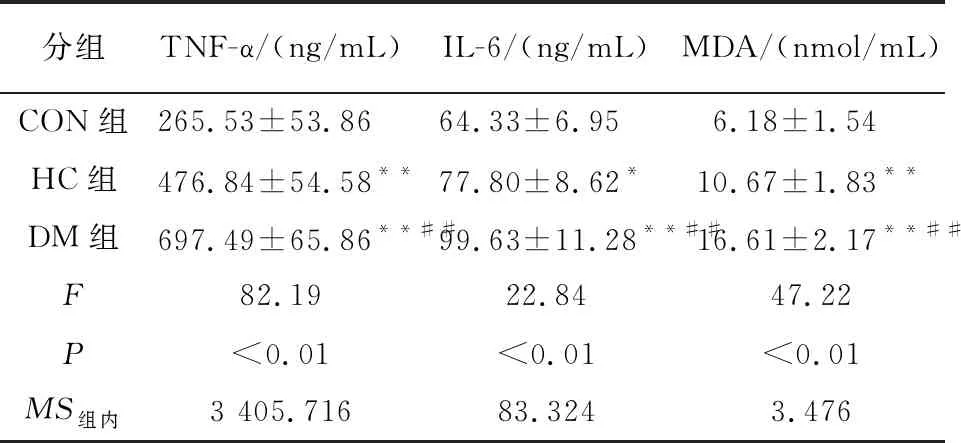
2.5 心肌组织RIPK1、RIPK3、MLKL的mRNA水平 与CON组比较,HC组的RIPK1及RIPK3的mRNA水平上调(P<0.05~P<0.01),MLKL无明显差异(P>0.05);DM组的RIPK1、RIPK3和MLKL的mRNA水平均高于CON组和HC组(P<0.01)(见表5)。
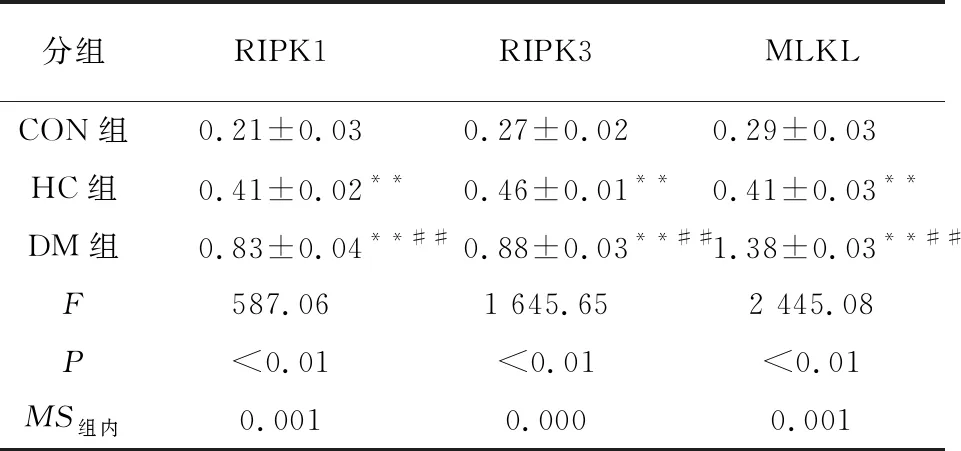
2.6 心肌组织RIPK1、RIPK3、MLKL的蛋白水平 HC组和DM组的RIPK1、RIPK3、MLKL的蛋白水平均较CON组上调(P<0.01);与HC组比较,DM组RIPK1、RIPK3、MLKL的蛋白水平均上调(P<0.01)(见图2、表6)。
2.7 心肌组织Masson染色 对各组大鼠心肌组织Masson染色结果显示,CON组大鼠心肌组织结构清晰,心肌细胞排列规则;DM组大鼠心肌组织纤维及细胞排列杂乱无序,细胞核散乱,细胞质淡染呈水样变,细胞间质内可见大量胶原纤维增生;而HC组则介于两者之间(见图3)。
3 讨论
目前我国糖尿病的控制情况不容乐观,据调查[9]显示,城市居民的糖尿病治疗比例为41.8%,而农村居民仅为27.6%。糖尿病主要表现为胰岛素分泌障碍及胰岛素抵抗而至血糖升高。DCM是由体内长期高糖环境及慢性低级别炎症和氧化应激等导致的心肌细胞或微血管损伤[10]。长期高糖环境加速脂质代谢紊乱,促进血管收缩,加剧内皮细胞受损,诱导内皮功能障碍,促动脉粥样硬化,是DCM发生发展的重要病理过程。在心脏功能正常的糖尿病病人的心肌细胞内已有脂质沉积,表明心肌细胞微环境改变发生在心衰之前[12]。罗清宇等[13]发现糖尿病模型组大鼠的心腔内径明显大于正常组。本实验发现,HC组小鼠较CON组小鼠外形略显肥胖,但BW无明显统计学意义,HW增加,LVEDD扩大,但心功能变化不明显,说明高脂喂养的大鼠虽然心功能没有较明显的恶化,但心腔结构及心肌负荷已发生明显改变,尤其是舒张末期内径增大,左心室前负荷增加,心脏做功增加,心功能尚属于代偿期,心肌收缩泵血尚能满足机体需要。本实验也通过复制糖尿病大鼠模型发现DM组大鼠较HC组大鼠,HW及BW减轻,LVEDD、LVESD和HW/BW增加,说明糖尿病大鼠明显处于消耗状态,BW减轻,心腔扩大,室壁应力增加,心脏离心性肥大。LVEF下降,泵血量占心腔充盈后总容积的比值明显下降,说明肥大的心肌及扩大的心腔已不能保证泵血量满足机体的需要,心脏泵血功能衰竭。Masson染色发现心肌细胞蓝染程度较高,心肌细胞损伤严重,出现修复性纤维化,说明高糖状态促进心肌细胞死亡,加速心衰进程。
分组TNF-α/(ng/mL)IL-6/(ng/mL)MDA/(nmol/mL)CON组265.53±53.86 64.33±6.95 6.18±1.54 HC组476.84±54.58**77.80±8.62*10.67±1.83**DM组697.49±65.86**##99.63±11.28**##16.61±2.17**##F82.1922.8447.22P<0.01<0.01<0.01MS组内3 405.71683.3243.476
q检验:与CON组比较*P<0.05,**P<0.01;与HC组比较##P<0.01
炎症伴随着组织巨噬细胞浸润和过度释放炎性细胞因子,包括TNF-α、IL-6和干扰素等。多种炎症因子参与2型糖尿病的发生、发展,包括糖尿病周围神经病变、肾损伤、牙周炎、糖尿病足等,与疾病转归及预后关系密切[14-16]。TNF-α与其同源受体TNF-1的配体结合诱导RIPK 1激酶激活,更是程序性坏死途径启动因素之一。IL-6由单核细胞、巨噬细胞和小胶质细胞刺激Toll样受体(TLRs)后迅速产生,结合TGF-β的IL-6优先诱导初始 CD4+T细胞分化为Th17细胞,参与慢性炎症疾病甚至癌症的发展[17-18]。氧化应激是指体内氧化物和抗氧化物失衡而造成的机体潜在损伤状态,MDA是脂质过氧化物的产物,因此可以间接反映体内氧自由基的代谢状况。故本研究选择大鼠血液中TNF-α、IL-6和MDA水平作为观察指标,观察糖尿病大鼠体内炎症因子及氧化应激水平。我们的研究表明,糖尿病大鼠的血清TNF-α、IL-6和MDA是高表达的,且CON组、HC组、DM组水平依次逐步增加,即长期高糖状态体内细胞氧化损伤增加,炎症因子释放,低水平的促炎状态又加重细胞氧化水平,最终使糖尿病大鼠心功能下降,产生较为典型的“三多一少”的症状。

分组RIPK1RIPK3MLKLCON组111HC组1.19±0.13**1.14±0.13*1.12±0.15 DM组 1.70±0.15**## 1.51±0.13**## 1.90±0.19**##F65.0636.9876.37P<0.01<0.01<0.01MS组内0.0120.0110.019
q检验:与CON组比较*P<0.05,**P<0.01;与HC组比较##P<0.01
RIPK1-RIPK3-MLKL通路起始于TNF-α及其相关配体结合,导致由RIPK1、TRADD、TRAF5、TRAF2、cIAPs组成的信号复合物Ⅰ的形成,RIPK1去泛素化促进复合物Ⅱb的组装,当caspases-8活性降低时(若caspases-8高表达则引起细胞凋亡),RIPK3磷酸化其特异性底物MLKL导致构象改变,暴露N-端死亡结构域,转移至细胞膜介导细胞死亡[19-21]。细胞Necroptosis不仅具有坏死的形态特点和自噬的活化,而且是耗能的,是可以调控的细胞死亡方式。既往实验[22]发现高糖诱导的损伤的肾小球脏层上皮细胞中RIPK 1、Caspase 3、RIPK 3和MLKL高表达,泛素羧基末端水解酶L1通过调节RIPK 1/RIPK 3通路的泛素化状态降低RIPK1、RIPK3和MLKL表达。Necroptosis核心成分在特定的肥胖相关组织中显著高表达,尤其是在糖尿病小鼠肝脏、脂肪组织和肌肉中检测到高水平的RIPK1和MLKL,RIPK3在脂肪组织中显著上调[23]。Necroptosis通路在缺血再灌注损伤后的心脏及脑内、肝细胞变性坏死、病毒性心肌炎、肿瘤增殖等多种病理状态中都是高表达的[24-26]。
分组RIPK1RIPK3MLKLCON组0.21±0.030.27±0.020.29±0.03 HC组0.41±0.02**0.46±0.01**0.41±0.03**DM组0.83±0.04**##0.88±0.03**##1.38±0.03**##F587.061 645.652 445.08P<0.01<0.01<0.01MS组内0.0010.0000.001
q检验:与CON组比较**P<0.01;与HC组比较##P<0.01
为了进一步观察RIPK1及其下游因子在糖尿病大鼠心肌中的水平,本研究采用了RT-PCR及Western Blotting 2种方法。结果发现,与CON组比较,HC组RIPK 1、RIPK 3的mRNA明显上调,MLKL的蛋白水平上调;DM组RIPK 1、RIPK 3和MLKL的mRNA和蛋白水平均明显上调。这和TNF-α、IL-6和MDA的表达水平一致。说明RIPK1介导的Necroptosis通路存在于在高糖状态下的心肌细胞中,糖尿病心肌中RIPK1-RIPK3-MLKL通路信号激活。这也和前人的研究[27]结果一致。较为遗憾的是本研究未能设置干预组,对比观察RIPK1等和IL-6等的表达量变化,这也是本研究小组将进行的进一步研究。
综上,本研究结果表明,高糖状态下的心肌细胞长期处于低水平的炎症状态,往往伴随着心肌纤维化及氧化应激,可能通过激活RIPK1介导的RIPK1-RIPK3-MLKL的信号通路而发生。Necroposis通路及其相关抑制剂在糖尿病方面具有很好的研究价值及应用前景。
