慢性阻塞性肺疾病模型大鼠核因子κB及肿瘤坏死因子α、白细胞介素8的表达水平及其意义▲
2020-05-08王会娟王昌明易玉芳
王会娟 王昌明 唐 灵 易玉芳
(桂林医学院附属医院全科医疗科,广西桂林市 541001,电子邮箱:wanghuij2004@163.com)
慢性阻塞性肺疾病(chronic obstructive pulmonary disease,COPD)是一种以持续气流受限为特征的疾病,可以预防和治疗。慢性缺氧、高碳酸血症和酸中毒可引起COPD患者出现肺血管重塑及广泛收缩而导致肺动脉高压。慢性炎症刺激还可导致血管内皮和平滑肌增生以及肺血管结构变化,例如狭窄、闭塞和纤维化,进一步导致肺血管重塑。吸烟、遗传因素、呼吸道感染和蛋白酶失衡与COPD的发病密切相关,但COPD的发病机制目前仍不清楚[1]。参与COPD发病机制的炎性细胞包括单核细胞、巨噬细胞和嗜中性粒细胞。当受到吸烟和感染等因素的刺激时,炎症细胞被激活并迅速释放细胞因子,而细胞因子进一步加剧炎症反应,诱导血管内皮细胞黏附分子的合成,进而损伤肺组织结构[1-2]。有研究表明,核因子κB (nuclear factor kappaB,NF-κB)参与机体的慢性炎症反应,白细胞介素(interleukin,IL)-8是促炎趋化因子[2],参与气道炎症的过程。肿瘤坏死因子α(tumor necrosis factor α,TNF-α)是一种多效性细胞因子,具有免疫调节以及促进细胞分化增殖、组织发育、血管生成炎症等作用[3],其可以增加肺血管反应性,并降低肺动脉平滑肌细胞前列腺素的产生,促使血小板活性因子诱导肺血管收缩[4]。本研究通过建立COPD大鼠模型及COPD并低氧大鼠模型,检测NF-κB、IL-8、TNF-α的表达水平,探讨上述指标在COPD发生发展中的作用机制。
1 材料与方法
1.1 实验动物与分组 无特定病原体级SD雄性大鼠 48只购自桂林医学院动物实验中心(动物许可证号:SYXK桂2013-0001),体重180~220g。采用随机数字表法将大鼠分为A组(正常对照组)、B组(COPD组)、C组(COPD并低氧组)、A1组(空白对照组)、B1组(COPD干预组)、C1组(COPD并低氧干预组),每组8只。
1.2 药物及试剂 脂多糖,吡咯烷二硫代氨基甲酸盐(pyrrolidine dithiocarbamate,PDTC)均购自美国Sigma公司(批号:Sigma-L2880、Sigma P8765 Ammonium),烤烟型香烟(甲天下牌)由广西卷烟厂出品(焦油量为14 mg烟气烟碱量:1.2 mg),钠石灰(批号:20160814)、无水氯化钙(批号:20160302)购自国药集团,医用氮气、医用氧气均由桂林医学院附属医院统一提供。鼠抗NF-κB p65购自美国 Santa Cruz公司(批号:sc-514451),β-肌动蛋白一抗(批号:AA132)、细胞裂解液(批号:P0013)、苯甲基磺酰氟(phenylmethanesulfonyl fluoride,PMSF)(批号:ST505)、辣根过氧化物酶体标记山羊抗小鼠IgG(批号:A0216)均购自江苏碧云天公司,大鼠IL-8酶联免疫吸附测定(enzyme-linked immunosorbent assay,ELISA)检测试剂盒购自上海西唐生物科技有限公司(批号:F15880),大鼠TNF-α ELISA检测试剂盒购自美国R&D公司(批号:MTA00B)。PDTC溶液的配置:用生理盐水将PDTC稀释成浓度为100 mg/mL溶液后备用。
1.3 主要仪器及设备 BP-221S电子分析天平(北京赛多利斯仪器系统有限公司);自制有机玻璃舱规格为0.91 m ×0.42 m×0.62 m;OX-100A数字测氧仪(上海隆拓仪器设备有限责任公司)。蛋白电泳及转膜仪(美国Bio-Rad公司),SensiCapture凝胶成像采集系统及 SensiAnsys凝胶图像分析系统均购自美国Media Cybernetics公司。
1.4 动物模型的构建及干预 (1)B组及B1组分别在实验第1、14天经气道注入脂多糖溶液,200 μg/次,之后将大鼠置于自制有机玻璃舱内熏烟1 h/d,5只烟/次,共6周,制作COPD大鼠模型;建模期间舱内小孔与外界相通,保证空气流通及压力与外界相同,舱内放置无水氯化钙及钠石灰,大鼠在舱内可自由进食;B1组在实验第15天开始直至处死每天予以PDTC溶液腹腔注射,剂量为100 mg/(kg·d);B组不做处理。(2)C组及C1组按照B组及B1组方法建立COPD模型,在实验的最后2周给予熏烟的同时,使用氮气调节氧浓度,使氧浓度稳定于18%,使用测氧仪监测氧浓度,低氧干预时间为8 h/d;C1组在实验第15天开始予以PDTC腹腔注射,用法同B1组;C组不做处理。(3)A组及A1组正常饲养。Al组于实验的第15天开始直至处死每天予腹腔注射与PDTC等体积的生理盐水;A组不做处理。
1.5 观察指标
1.5.1 病理组织学:实验6周后,大鼠经1%戊巴比妥钠(40~55 mg/kg)腹腔麻醉后,行右心室采血后,用组织剪分离肺脏。取大鼠右肺中叶,于肺内注射 10%中性福尔马林至肺膨胀后结扎肺门,标本浸泡于福尔马林中固定24 h,于右中叶距肺门2~3 mm处取材,取材后常规石蜡包埋、切片;行苏木精-伊红 (hematoxylin-eosin,HE)染色及维多利亚蓝+Van-Gieson染色,观察大鼠病理组织学情况。(1)气道重构:在200倍镜视野下任选5个直径≤1 100μm(管腔最短径/最长径≥0.7)的细支气管,测量其管壁面积、气管总面积、细支气管外径、管壁厚度;然后计算管壁面积和气管总面积比值即气管中膜面积(用MA%表示),以及管壁厚度与气管外径比值即气管中膜厚度(用MT%表示)。(2)肺血管重塑:在200倍镜下测定直径<100 μm的肺腺泡内的肺动脉总数,将其分类,分别计算肌性动脉、部分肌性动脉、非肌性动脉构成比。在400倍镜下对50~100 μm的肌性动脉进行图像分析,计算血管管壁面积与血管总面积比即肺动脉WA%,以及血管管壁厚度与血管外径比即肺动脉WT%。
1.5.2 NF-κB p65蛋白表达水平:取右肺前叶及后叶组织,液氮中碾磨,按总蛋白提取试剂盒(上海贝博生物科技有限公司,批号:310003)操作步骤提取组织总蛋白,即碾磨后肺组织粉末加入蛋白提取液、PMSF、蛋白酶抑制剂,充分混匀后冰上裂解30 min,12 000 r/min,4℃离心15 min,取上清(即组织总蛋白)。将上清液移入另一离心管中保存置于-80℃待检。待检蛋白样品用二喹啉甲酸法进行蛋白定量,操作步骤按照二喹啉甲酸蛋白定量试剂盒(碧云天生物科技有限公司,批号:P0012S)说明书进行。经12% 聚丙烯酰胺凝胶电泳分离蛋白后,转移至聚偏二氟乙烯膜上,5%脱脂奶粉封闭1 h,用TBST充分洗涤,加入抗NF-κB p65单克隆抗体(稀释度1 ∶800),于室温孵育2 h;充分洗膜后加入二抗山羊抗鼠IgG(稀释度为1 ∶1 000),于室温孵育1 h;TBST充分洗涤后,进行显影和定影,于凝胶成像分析系统分析胶片,以β-肌动蛋白为内参(一抗稀释度1 ∶500,二抗稀释度1 ∶1 000),以NF-κB p65与β-肌动蛋白比值作为目的蛋白相对表达水平。
1.5.3 肺泡灌洗液及血清中TNF-α、IL-8水平:从心脏取血约5 mL,放入离心管,3 000 r/min离心15 min,取上层血清于-70℃保存。大鼠肺脏用组织剪离体后以丝线结扎右肺支气管,采用0.1 mol/L的磷酸缓冲盐溶液灌洗左肺3次,每次2 mL,获得肺泡灌洗液(bronchoalveolar lavage fluid,BALF)后,1 500 r/min离心10 min,取上清液置于-80℃保存。采用ELISA法检测TNF-α、IL-8的表达水平,按试剂盒说明书进行操作,于酶标仪的450 nm波长处读取各孔A值。
1.6 统计学分析 采用SPSS 17.0软件进行统计学分析。计量资料以(x±s)表示,多组样本间均数比较采用单因素方差分析,两两比较采用SNK-q检验,以P<0.05为差异具有统计学意义。
2 结 果
2.1 肺组织病理学 A组与A1组镜下未见炎症细胞浸润,管壁完整,为正常气道结构。B组镜下可见典型的COPD病变表现,包括气道管壁有炎症细胞浸润,气道上皮细胞增生,肺气肿明显,并可见肺大疱形成,与B组比较,Bl组炎症细胞浸润减少,肺气肿、肺大疱有改善。C组镜下观察可见典型的COPD并低氧的病理表现,包括气道管壁大量炎症细胞浸润,局部可见肌层断裂及肺泡壁断裂,平滑肌增生明显,与C组比较,C1组炎症浸润、肌层断裂、肺泡壁断裂现象均减少。
2.2 6组大鼠气道及肺血管重构指标比较 除部分肌性动脉率外,6组大鼠其他气道及肺血管重构指标差异均有统计学意义(均P<0.05)。与A组比较,B、C组肌性动脉率以及肺动脉MA%、肺动脉MT%均增高,而非肌性动脉率降低(均P<0.05),但B组和C组间差异无统计学意义(P>0.05);B1组、C1组的肌性动脉率、肺动脉MA%、肺动脉MT%分别低于B组及C组,而非肌性动脉率分别高于B组及C组(均P<0.05)。C组的气管WA%、气管WT%均高于A组(均P<0.05),但B组与A组间差异无统计学意义(P>0.05);C1组气管WA%、气管WT%均低于C组(均P<0.05),但B1与B组间差异无统计学意义(P>0.05)。见表1。
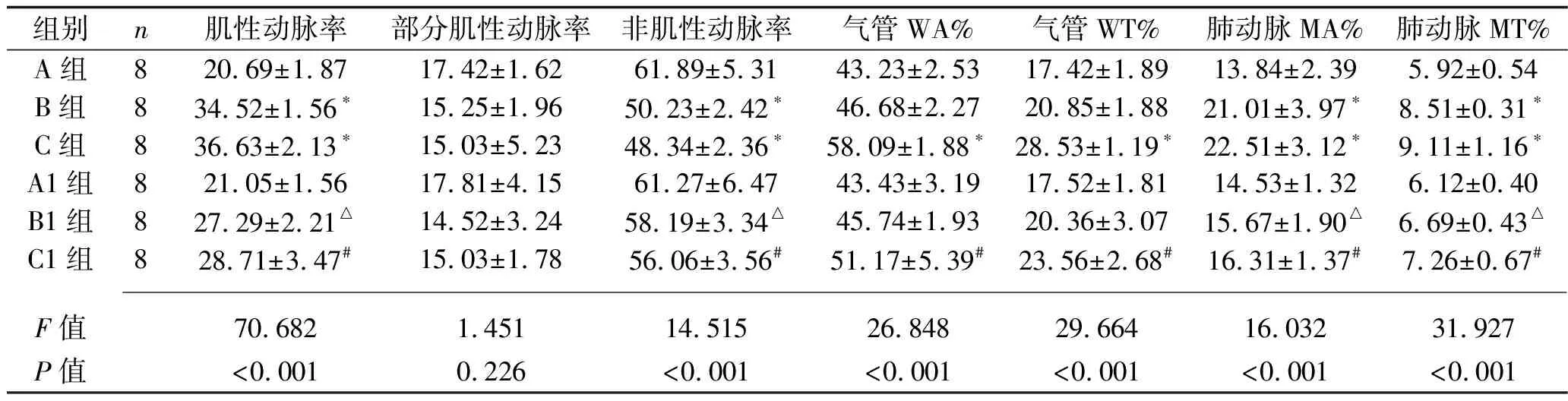
表1 6组肺血管及气道重构指标比较(x±s)
注:与A组比较,*P<0.05;与B组比较,△P<0.05;与C组比较,#P<0.05。
2.3 6组肺组织NF-κB p65蛋白表达水平比较 与A组比较,B、C组NF-κB p65蛋白相对表达水平增高,且C组高于B组(均P<0.05);B1组、C1组的NF-κB p65蛋白相对表达水平分别低于B组、C组(均P<0.05)。见表2。
2.4 6组大鼠BALF及血清中IL-8,TNF-α表达水平比较 与A组比较,B组、C组血清及BALF中IL-8、TNF-α水平增高,且C组均高于B组(均P<0.05);B1、C1组血清及BALF中IL-8、TNF-α水平分别低于B组、C组(均P<0.05)。 见表2。
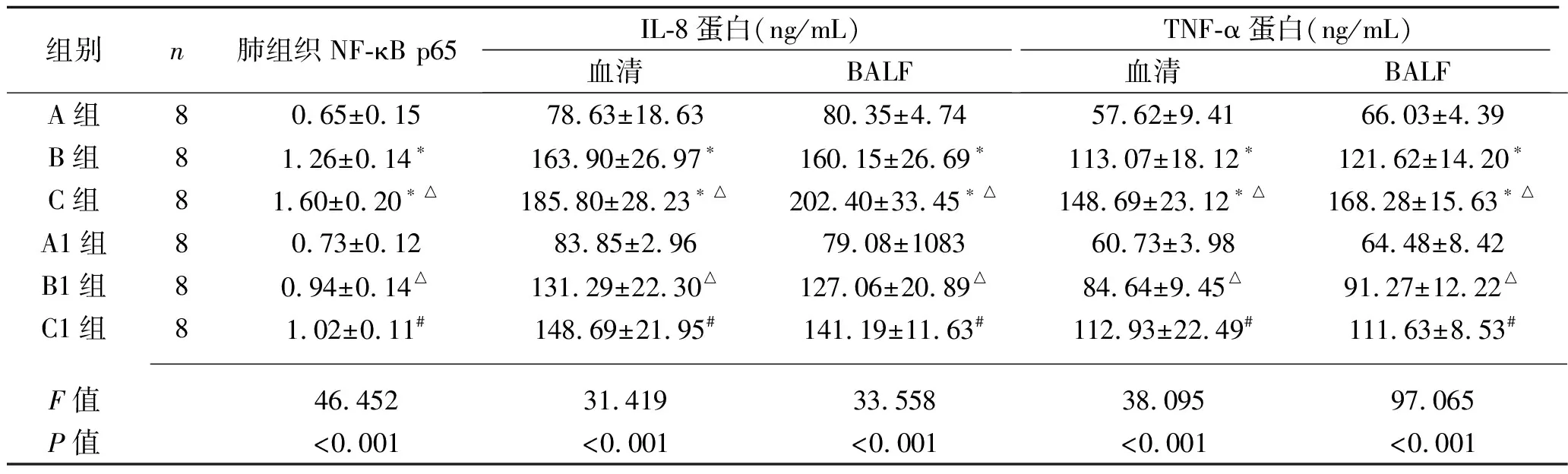
表2 6组肺组织NF-κB p65蛋白、BALF及血清中IL-8和TNF-α蛋白表达水平比较(x±s)
注:与A组比较*P<0.05;与B组比较,△P<0.05;与C组比较,#P<0.05。
3 讨 论
COPD 的病理特征是累及气道、肺实质以及肺血管的慢性炎症和气道重构[5]。随着COPD的进展,可能会出现肺血管重建和肺动脉高压,进一步引起肺心病。细胞因子在COPD的发病过程中起关键作用,以TNF-α、IL-8为主要细胞因子导致的气道炎症被认为是COPD发病的重要机制之一。
本实验通过叠加吸烟、感染、低氧三大致病因素成功制备COPD及COPD并低氧动物模型,分别模拟COPD大鼠模型合并不同程度的肺血管重构状态。结果显示,B组COPD大鼠气道阻力增高,管腔内炎细胞聚集及黏液蓄积,管壁内大量炎症细胞浸润,柱状上皮杯状细胞增生明显,平滑肌层增厚,黏膜下及外膜胶原纤维增生,部分肌层断裂,肺气肿形成,部分肺泡壁断裂形成肺大疱,肌性动脉百分比增高;C组COPD并低氧大鼠病理改变与COPD组相似,但平滑肌层增厚更明显。上述模型代表COPD肺血管重构的不同阶段,其中COPD模型代表早期阶段,表现为肌性动脉百分比增多;COPD并低氧模型代表肺血管重构进展阶段,此时不仅腺泡内肌性动脉百分比增高,且肺动脉中膜厚度增加,WA%、WT%比值增高。相关病理学改变均与人类COPD的病理特征吻合。
NK-κB通常以p50/p65异源二聚体的形式存在,该二聚体也是其主要活性形式。其中,p50含核定位信号,与DNA直接结合,p65含转录活化区域,参与基因转录的起始调节,具有转录激活功能。在正常情况下,由p50/p65二异聚体与抑制性蛋白结合的成三聚体,处于无活性状态。但多种刺激诱导可活化NF-κB,NF-κB的活化会引起一系列基因表达,包括炎症刺激相关的细胞因子和生长因子,经过NF-κB诱导产生的蛋白,如TNF-α、IL-8等,又可活化NF-κB,最终使炎症发生级联化反应。多项临床研究结果均表明COPD患者的血清TNF-α和IL-8水平高于正常健康人[6-7]。本研究结果显示,COPD大鼠及COPD并低氧大鼠NF-κB p65蛋白、TNF-α、IL-8表达水平升高,且COPD并低氧的大鼠更明显。这提示TNF-α、IL-8参与COPD发生和发展过程,随着疾病的发展,相关炎症因子水平也升高,而NF-κB可能起到上游调控的作用[8]。
还有研究表明,NF-κB不仅在炎症因子网络中起重要的调节作用,还参与肺血管重构、肺动脉对刺激因子的收缩反应、肺动脉平滑肌细胞的增殖及凋亡等过程。NF-κB还可通过炎症介质来干预一氧化氮、内皮素1、血管内皮生长因子、成纤维细胞生长因子、转化生长因子β等的调节,进而影响肺血管重构[9]。本研究结果显示,与A组比较,B、C组肺动脉MA%、MT%、肌性动脉率均增高,且肺组织NF-κB p65蛋白的表达水平亦升高(P<0.05),提示随着气道、肺血管重构的发展,炎症因子的表达也呈现逐渐增加的趋势。说明炎症反应与COPD的气道、肺血管重构密切相关。
PDTC是NF-κB特异性抑制剂,其可抑制NF-κB的激活,进而抑制炎症因子的表达[10]。本研究结果显示,与对应模型组比较,B1组、C1组NF-κB p65蛋白表达、TNF-α和IL-8水平均降低,且气管及血管重塑指标(肺动脉MA%及肺动脉MT%、肌性动脉率)均下降(P<0.05),提示抑制NF-κB p65蛋白的表达,可直接下调TNF-α及IL-8的表达水平,并改善气管及肺血管重构。因此,PDTC对改善肺部炎症及肺血管重构,延缓COPD病程进展,或有一定疗效。
综上所述,COPD大鼠的NF-κB及TNF-α、IL-8等炎症因子的表达均上调,NF-κB可能是通过调节TNF-α、IL-8等早期炎症介质的表达从而参与COPD大鼠肺血管重塑、肺动脉高压的形成过程。
