MicroRNA regulatory pattern in spinal cord ischemiareperfusion injury
2020-04-29ZhiGangLiuYinLiJianHangJiaoHaoLongZhuoYuanXinXiaoYuYang
Zhi-Gang Liu, Yin Li, Jian-Hang Jiao, Hao Long, Zhuo-Yuan Xin, Xiao-Yu Yang,
1 Department of Orthopedics, The Second Hospital of Jilin University, Changchun, Jilin Province, China
2 School of Public Health, Jilin University, Changchun, Jilin Province, China
3 Pain Clinic, The Sixth Affiliated Hospital of Xinjiang Medical University, Urumqi, Xinjiang Uygur Autonomous Region, China
4 The Key Laboratory of Zoonosis Search, Chinese Ministry of Education, College of Basic Medicine, Jilin University, Changchun, Jilin Province,China
Abstract After spinal cord injury, dysregulated miRNAs appear and can participate in inflammatory responses, as well as the inhibition of apoptosis and axon regeneration through multiple pathways. However, the functions of miRNAs in spinal cord ischemia-reperfusion injury progression remain unclear. miRCURY LNATM Arrays were used to analyze miRNA expression profiles of rats after 90 minutes of ischemia followed by reperfusion for 24 and 48 hours. Furthermore, subsequent construction of aberrantly expressed miRNA regulatory patterns involved cell survival, proliferation, and apoptosis. Remarkably, the mitogen-activated protein kinase (MAPK) signaling pathway was the most significantly enriched pathway among 24- and 48-hour groups. Bioinformatics analysis and quantitative reverse transcription polymerase chain reaction confirmed the persistent overexpression of miR-22-3p in both groups. These results suggest that the aberrant miRNA regulatory network is possibly regulated MAPK signaling and continuously affects the physiological and biochemical status of cells, thus participating in the regulation of spinal cord ischemia-reperfusion injury. As such, miR-22-3p may play sustained regulatory roles in spinal cord ischemia-reperfusion injury. All experimental procedures were approved by the Animal Ethics Committee of Jilin University, China[approval No. 2020 (Research) 01].
Key Words: gene regulatory networks; microarray analysis; microRNA; miR-22-3p; mitogen-activated protein kinase signaling pathway; nerve regeneration; neural regeneration; spinal cord ischemia-reperfusion injury; transcriptome
Introduction
Spinal cord ischemia-reperfusio n injury (SCIRI), one of the most serious secondary spinal cord injuries, remains an unpredictable and devastating complication in clinical practice, with an incidence of 2-40% (Coselli et al., 2000, 2002).A cascade of secondary damage may be induced by SCIRI,such as neuronal lesions, which leads to further functional loss and behavioral impairments, as well as paraplegia (Bell et al., 2015; Fang et al., 2015). Importantly, there are no efficacious drugs or therapeutic approaches for SCIRI. Thus,many patients suffer substantial physical and psychological consequences. To date, the molecular mechanisms of SCIRI remain unclear, so it is difficult to develop novel drugs or treatments. Thus, it is urgent to identify the molecular mechanisms that underlie the pathogenesis of SCIRI.
Aberrant gene expression is associated with the development of numerous diseases and may involve many types of regulatory molecules. MicroRNA (miRNA) is an important kind of regulator that functions in RNA silencing and can subsequently regulate gene expression at the post-transcriptional level (Ambros, 2004; Bartel, 2004). miRNAs that remit inflammatory damage to the blood-spinal cord barrier, such as miR-27a, may have essential roles in the progression of SCIRI (Li et al., 2015). Transcription factors (TFs), another important regulator of gene expression at the transcriptional level (Yang et al., 2013), are proteins coded by specific genes that control the transcription rate of other coding genes.Interactions between miRNAs and TFs provide complex information in molecular regulatory patterns. Therefore, alterations in miRNA-TF gene expression regulatory networks may result in the development of SCIRI. Unfortunately, the aberrant miRNA-TF gene regulatory patterns in SCIRI remain unclear. Thus, a systemic analysis of altered miRNA-TF co-regulatory patterns would facilitate understanding of the molecular mechanisms of SCIRI.
This study aimed to analyze miRNA expression profiles and construct a miRNA regulatory pattern in a SCIRI rat model. miRNA expression profiles were analyzed using a miRCURY®LNA®Array (Qiagen, Hilden, Germany). The targets of aberrantly expressed miRNAs were subsequently predicted by integrating the data in three miRNA information databases. Altered miRNA-TF regulatory patterns were constructed based on the Transcriptional Regulatory Element Database (http://rulai.cshl.edu/tred). Gene ontology(GO; http://www.geneontology.org) annotation and Kyoto Encyclopedia of Genes and Genomes (KEGG) pathway enrichment (https://www.kegg.jp) were performed to provide a preliminary analysis of the function of aberrant miRNA-TF patterns in SCIRI, and the main findings were validated by quantitative reverse transcription-polymerase chain reaction(qRT-PCR). The ultimate goal of this study was to provide important clues for future investigations into the molecular mechanisms of SCIRI and facilitate the development of novel therapeutic approaches for this devastating injury.
Materials and Methods
Animals
All experimental procedures were approved by the Animal Ethics Committee of Jilin University, China [approval No.2020 (Research) 01]. The experimental procedure followed the United States National Institutes of Health Guide for the Care and Use of Laboratory Animals (NIH Publication No. 85-23, revised 1996). Twenty-four male mature Sprague-Dawley rats weighing 220-280 g were purchased from Beijing HFK Bioscience Co., Ltd. [Beijing, China;Animal License No. SCXK (Jing) 2009-0004]. All rats were raised at the School of Public Health at Jilin University,housed in standard cages, and neurologically intact prior to anesthesia. Animals were individually housed after surgery.
SCIRI rat models
To establish SCIRI rat models, 24 clean healthy adult male Sprague-Dawley rats were anesthetized by intraperitoneal injection with 4% sodium pentobarbital (Solarbio Life Science, Beijing, China) at a dose of 50 mg/kg. SCIRI was induced by abdominal aorta occlusion for 90 minutes. Following anesthetization, the abdominal aorta was exposed using a cervicothoracic approach. The abdominal aorta was subsequently cross-clamped between the left renal artery and origin of the right renal artery using non-invasive arterial clips. The occlusion situation was confirmed by detecting beating of the distal abdominal aorta (if clamped successfully, the beating would stop and the color of the left renal would change to dark red). After 90 minutes of ischemia, the arterial clips were removed and reperfusion was allowed to occur for 24 (24-hour group,n= 8) or 48 (48-hour group,n= 8) hours. Sham-operated rats (surgery without abdominal aortic clamp) were used as the control group (n= 8). The Basso Beattie Bresnahan locomotor rating scale was used to evaluate SCIRI rat models (Basso et al., 1995). Sham-operated rats underwent the same procedure without abdominal aortic occlusion.
RNA isolation, microarray hybridization, and signal scanning
Following the establishment of SCIRI rat models, total RNA in lesion tissues was extracted using TRIzol reagent (Invitrogen, Carlsbad, CA, USA) according to the manufacturer’s instructions, and then purified with an RNeasy Mini kit (Qiagen). RNA purity and concentration were detected using an UV2800 ultraviolet spectrophotometer (UNICO National,Fairfield, NJ, USA), which required concentrations ranging between 100 ng/mL and 1 mg/mL, anA260nm/A280nmratio between 1.8 and 2.0, andA260nm/A230nmratio greater than 1.8.
Following the manufacturer’s instructions, miRCURY LNA Array v.16.0 software (Qiagen) was used to profile differentially expressed miRNAs in lesion and normal tissues. Briefly, the extracted total RNA was labeled with a miRCURY Array Power Labeling kit (Qiagen). Labeled total RNAs were subsequently hybridized to the miRCURY LNA Array by incubation at 56°C and rotated at 2 rpm overnight.Fluorescence intensities of the miRCURY LNA Array were scanned using a GenePix 4000B scanner and analyzed with GenePix Pro v.6.0 software (Axon Instruments, Foster City,CA, USA).
MiRNA-TF co-regulatory network analysis
After miRNA expression profile data were obtained, the aberrantly expressed miRNAs with fold changes over 1.5 and P-values less than 0.05 were identified. Regulatory interactions among these differentially expressed miRNAs and target genes were predicted via data integration using miRbase(www.mirbase.org/) (Griffiths-Jones et al., 2006), miRDB(http://mirdb.org) (Wang, 2008), and miRanda (http://www.microrna.org/microrna/home.do) (Betel et al., 2008). TFs were identified based on information provided by the Transcriptional Regulatory Element Database (Zhao et al., 2005).miRNA-TF regulatory patterns were constructed by overlapping these two results, and subsequently visualized using the free software Cytoscape v.3.2.0 (Shannon et al., 2003).
Functional annotation and KEGG pathway enrichment analysis
MiRNA-TF regulatory patterns were identified using The Database for Annotation, Visualization, and Integrated Discovery (https://david.ncifcrf.gov/) (Dennis et al., 2003) and tools in KEGG (Kanehisa and Goto, 2000) to conduct functional annotation and KEGG pathway enrichment analyses.Significantly enriched KEGG pathways were sorted byPvalues less than 0.05.
Quantitative reverse transcription-polymerase chain reaction
The miRNA-TF regulatory patterns identified persistent overexpression of miR-22-3p during SCIRI. qRT-PCR was used to verify expression trends of miR-22-3p. For qRT-PCR,5 mg of total RNA extracted from lesion and normal spinal cord tissues was reverse-transcribed into cDNA using a firststrand cDNA synthesis kit (Takara, Tokyo, Japan). miRNA expression of rno-miR-22-3p was detected using SYBR Premix Ex Taq (Takara) and an Applied Biosystems 7300 Fast Real-Time PCR System (Foster City, CA, USA). Relative miRNA expression was normalized to U6 expression levels using the comparative 2-ΔΔCtmethod (ΔCt = CtTarget- CtU6,ΔΔCt = ΔCtLesion- ΔCtNormal). Primers were designed using Primer Premier software, version 6.0 (Primer Biosoft, Palo Alto, CA, USA) and Primer-BLAST (https://www.ncbi.nlm.nih.gov/tools/primer-blast/). qRT-PCR data were analyzed using Prism Version 5.0 software (GraphPad, San Diego, CA,USA). Rno-miR-22-3p specific primers were 5′-GGT TAA GCT GCC AGT TGA A-3′ (forward, 19 bp) and 5′-CAG TGC GTG TCG TGG AGT-3′ (reverse, 18 bp) (Kang Chen Bio-tech, Shanghai, China). Reaction conditions included a denaturation step at 95°C for 10 minutes, followed by 40 cycles of 95°C for 10 seconds and 60°C for 60 seconds. Primers used to amplify U6 were 5′-GCT TCG GCA GCA CAT ATA CTA AAA T-3′ (forward, 25 bp) and 5′-CGC TTC ACG AAT TTG CGT GTC AT-3′ (reverse, 23 bp) (Kang Chen Bio-tech).
Statistical analysis
Measurement data are expressed as the mean ± SD and were analyzed using R version 3.1.2 software (https://www.r-project.org). All variables were normally distributed, and groups were compared with one-way analysis of variance followed by the least significant differencepost hoctest.P-values < 0.05 were considered statistically significant.
Results
Differentially expressed miRNAs in the lesion region of SCIRI rat models
To identify potential roles of miRNAs in SCIRI, miRNA expression profiles were first detected in SCIRI lesion regions at 24 and 48 hours after reperfusion for comparison with sham-operated tissues. The results demonstrated that among the 695 detected rat miRNAs, at 24 hours, 13 miRNAs were aberrantly expressed, including 12 upregulated miRNAs and 1 downregulated miRNA (Figure 1A and Table 1). At 48 hours, 105 miRNAs were differentially expressed, including 44 upregulated miRNAs and 61 downregulated miRNAs (Figure 1B and Table 2). Rno-miR-22-3p was significantly upregulated at both 24 and 48 hours after reperfusion (Figure 1C).
Aberrant putative miRNA-TF regulatory patterns in SCIRI rat models
To construct a landscape of miRNA-TF regulatory patterns in SCIRI, the targets of all aberrantly expressed miRNAs were predicted using the miRNA databases miRbase,miRDB, and miRanda (Additional Table 1). General miRNA-TF regulatory patterns (Additional Table 2) were sequentially built based on rat TF information provided by the Transcriptional Regulatory Element Database (Additional Table 3). These networks indicated systemic miRNA-TF regulatory patterns. Furthermore, the regulatory strategies of miRNAs were substantially more complex at 48 hours compared with 24 hours after reperfusion (Figure 2 and Additional Figures 1 and 2). miRNA-TF regulatory patterns indicated the involvement of TFs associated with cell proliferation, survival, and apoptosis, such as Egr2, Stat3, Ets1,Tp53, and Hif-1α. These findings suggest that apoptosis and inflammation may represent important biological processes involved in the progression of SCIRI.
Functional annotation and KEGG pathway enrichment analysis
To determine the function of putative miRNA-TF regulatory patterns, GO gene functional annotation was conducted for all predicted miRNA-target genes. Figure 3 shows the top 20 significant GO annotation terms that exhibited a significant difference between 24 and 48 hours after reperfusion. Inter-estingly, subsequent KEGG enrichment analysis indicated that the mitogen-activated protein kinase (MAPK) signaling pathway was most significantly enriched in both 24- and 48-hour SCIRI groups (Tables 3 and 4). This finding indicates that the MAPK signaling pathway may play a critical role during the development of SCIRI and, therefore, represent a potential therapeutic target. Thus, potential correlations were analyzed between differentially expressed miRNAs and the MAPK signaling pathway. All associated miRNA-target genes were mapped to the MAPK signaling pathway (Figure 4A), and ideographs were created to demonstrate how these differentially expressed miRNAs regulate MAPK signal transduction processes via MAPK/ERK1/2 (Figure 4B),MAPK/p38 (Figure 4C), and MAPK/c-JNK (Figure 4D).
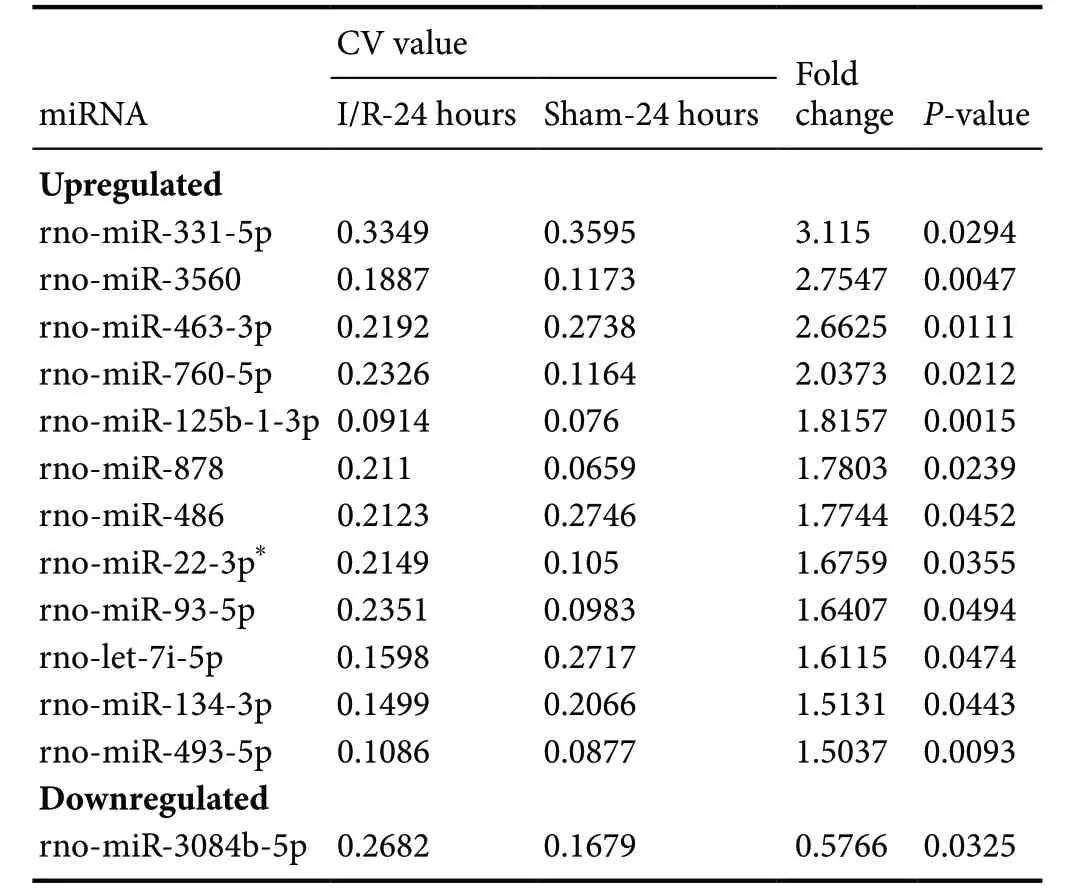
Table 1 Differentially expressed miRNAs in spinal cords among SCIRI model rats at 24 hours
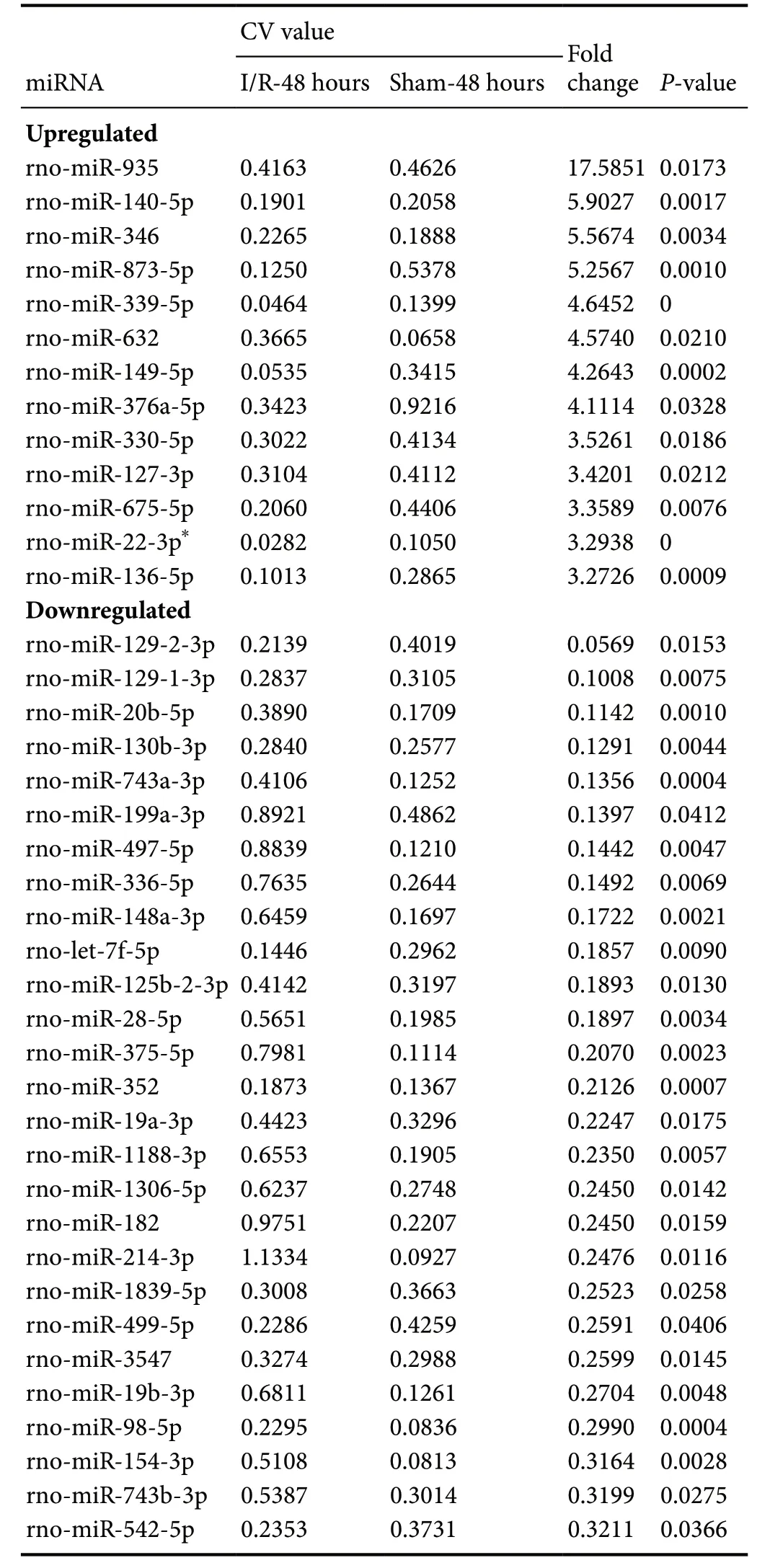
Table 2 Differentially expressed miRNAs in spinal cords among SCIRI model rats at 48 hours
Verification of findings by qRT-PCR
miRNA-TF regulatory patterns identified in this study were based on miRNA expression profile data. Therefore, qRTPCR was used to validate rno-miR-22-3p, which was significantly upregulated at both 24 and 48 hours after reperfusion in spinal cord lesion tissues compared with control tissues.qRT-PCR findings were consistent with miRNA expression profile data (Figure 5).
Discussion
SCIRI triggers a cascade of molecular alteration events that can lead to neuronal cell death (Juurlink and Sweeney, 1997;White et al., 2000). Inflammation, reactive oxygen species,excitotoxicity (Sattler and Tymianski, 2001), and apoptosis(Gottlieb et al., 1994; Chopp and Li, 1996) are the primary mechanisms involved in SCIRI. To better understand the molecular mechanisms of SCIRI, the current study identified all differentially expressed miRNAs in a rodent model.miRNAs are delicate gene expression regulators with special abilities to regulate the translation and stability of their targets by inhibiting translational activity and promoting target mRNA cleavage (O’Connell et al., 2012; Ksiazek-Winiarek et al., 2013). In view of this fundamental function of miRNAs and their interactions with TFs, miRNAs may act as key regulators in many biological processes in SCIRI, such as apoptosis, cell proliferation, responses to reactive oxygen species,and inflammatory responses. Thus, miRNAs represent potential therapeutic targets for SCIRI.
The current novel findings indicated that miR-22-3p was upregulated at both 24 and 48 hours after reperfusion. miR-22-3p was obviously increased at 48 hours after reperfusion.In myocardial ischemia-reperfusion injury, miR-22 reportedly exhibited anti-apoptosis effects in rats via the downregulation of CREB-binding protein expression, which may decrease the acetylation levels of p53, Bax, and p21 (Yang et al., 2014). MiR-22 overexpression elicits neuroprotection through its targets in general anti-apoptosis signaling pathways, including MAPK/p38 and p53 (Jovicic et al., 2013;Yang et al., 2014). Other differentially expressed miRNAs identified at 24 or 48 hours after reperfusion exhibited a broad range of functions. For example, miR-29c overexpression at 48 hours may exhibit pro-apoptosis effects via the suppression of anti-apoptotic gene expression products (Ye et al., 2010; Armato et al., 2012). miR-451 upregulation at 48 hours may decrease cardiomyocyte death in simulated ischemia-reperfusion injuryin vitro, as it increases COX-2 expression to protect against cardiac injury (Gray et al., 1990;Penna et al., 2008; Inserte et al., 2009; Zhang et al., 2010).Furthermore, miR-199a targets hypoxia-inducible factor 1α,which in turn, stabilizes the pro-apoptotic TF p53. Thus, the observed decrease of miR-199a at 48 hours in this study may prevent ischemia-reperfusion injury in the heart, kidney, and any other tissues (Rane et al., 2009; Wang et al., 2014; Bi et al., 2015; Nanayakkara et al., 2015). However, the functions of many other differentially expressed miRNAs identified in this study have not been previously reported and, thus, require further investigation.

Table 3 KEGG pathway enrichment analysis of the predicted miRNA-target genes in the spinal cords of rats 24 hours after ischemia-reperfusioin injury
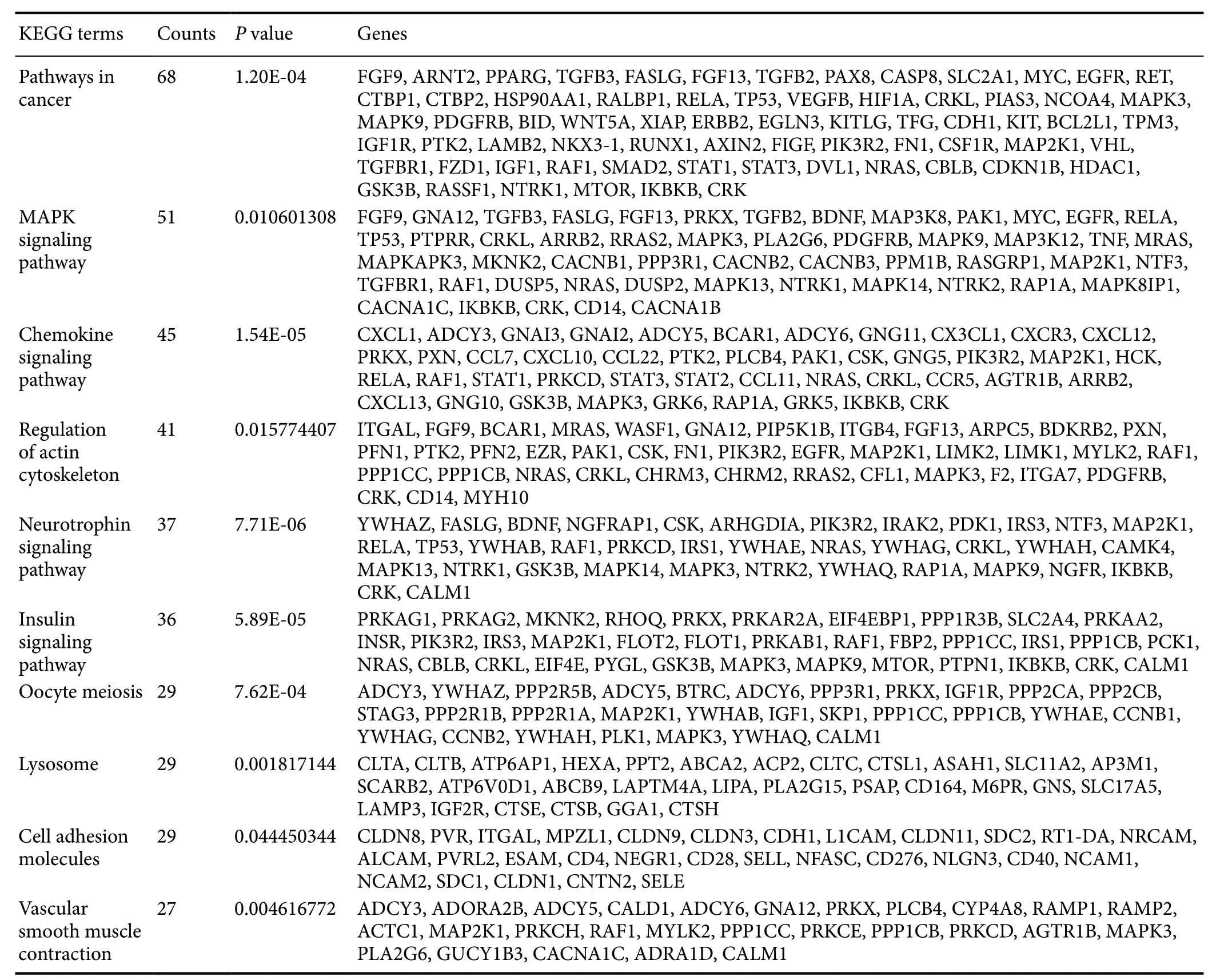
Table 4 KEGG pathway enrichment analysis of the predicted miRNA-target genes in the spinal cords of rats 48 hours after ischemia-reperfusioin injury
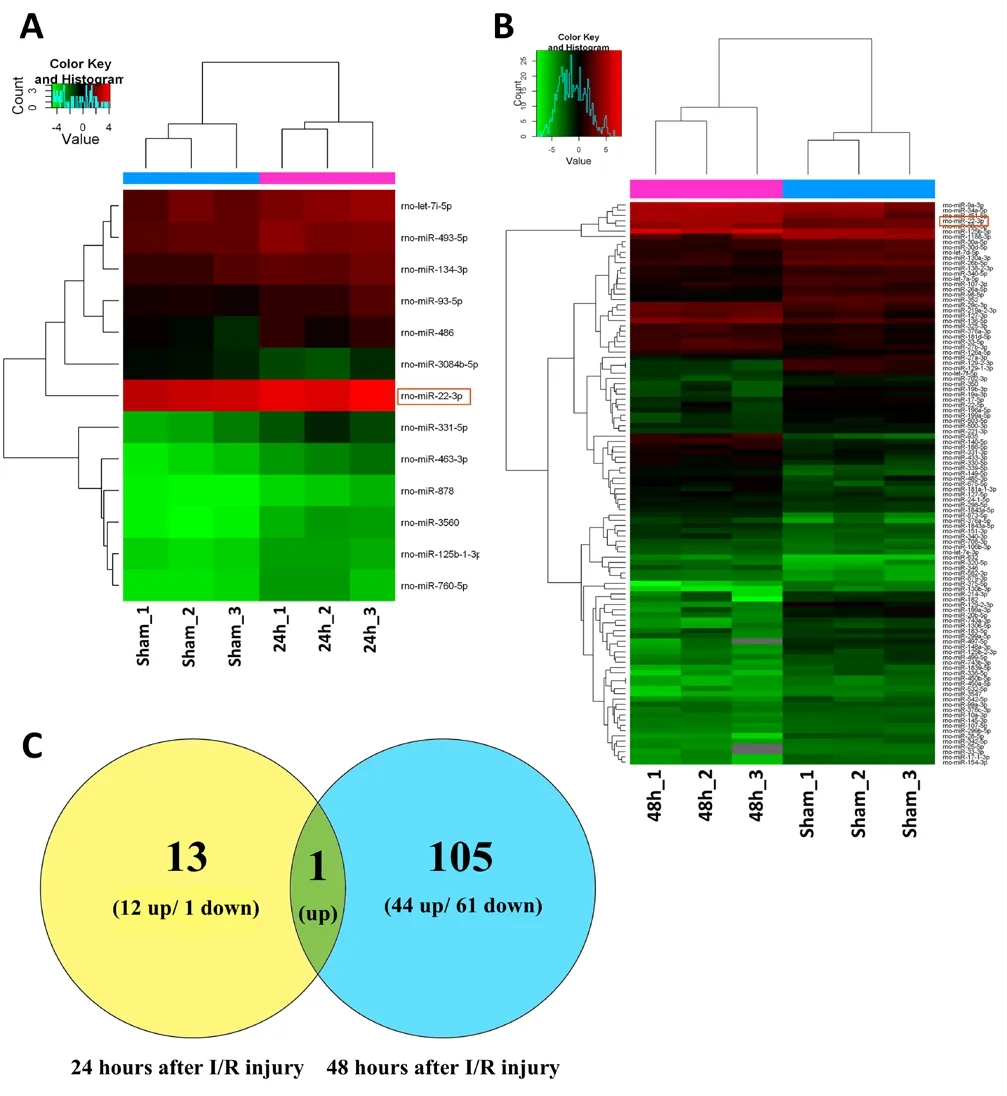
Figure 1 Cluster analyses of altered miRNAs after SCIRI.
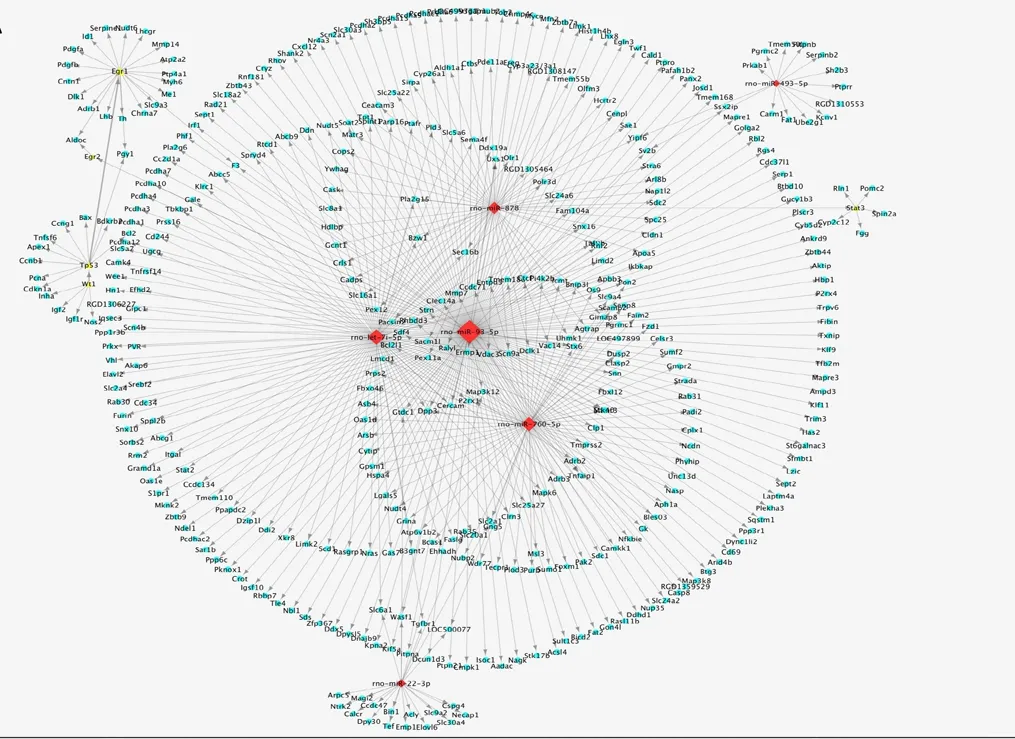
Figure 2 Putative miRNA-TF regulatory patterns in spinal cord ischemia-reperfusion injury.
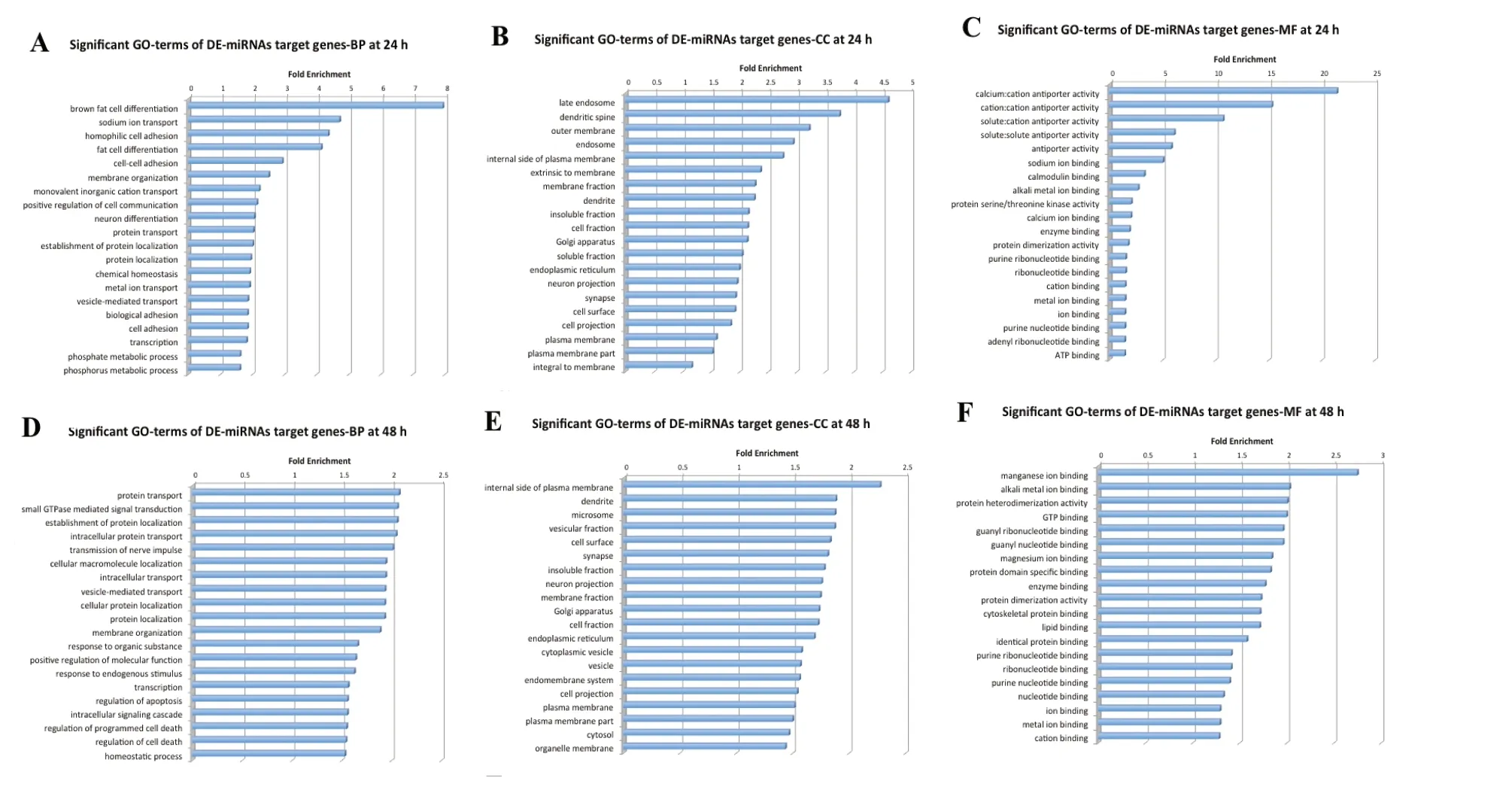
Figure 3 GO annotation of all predicted target genes.
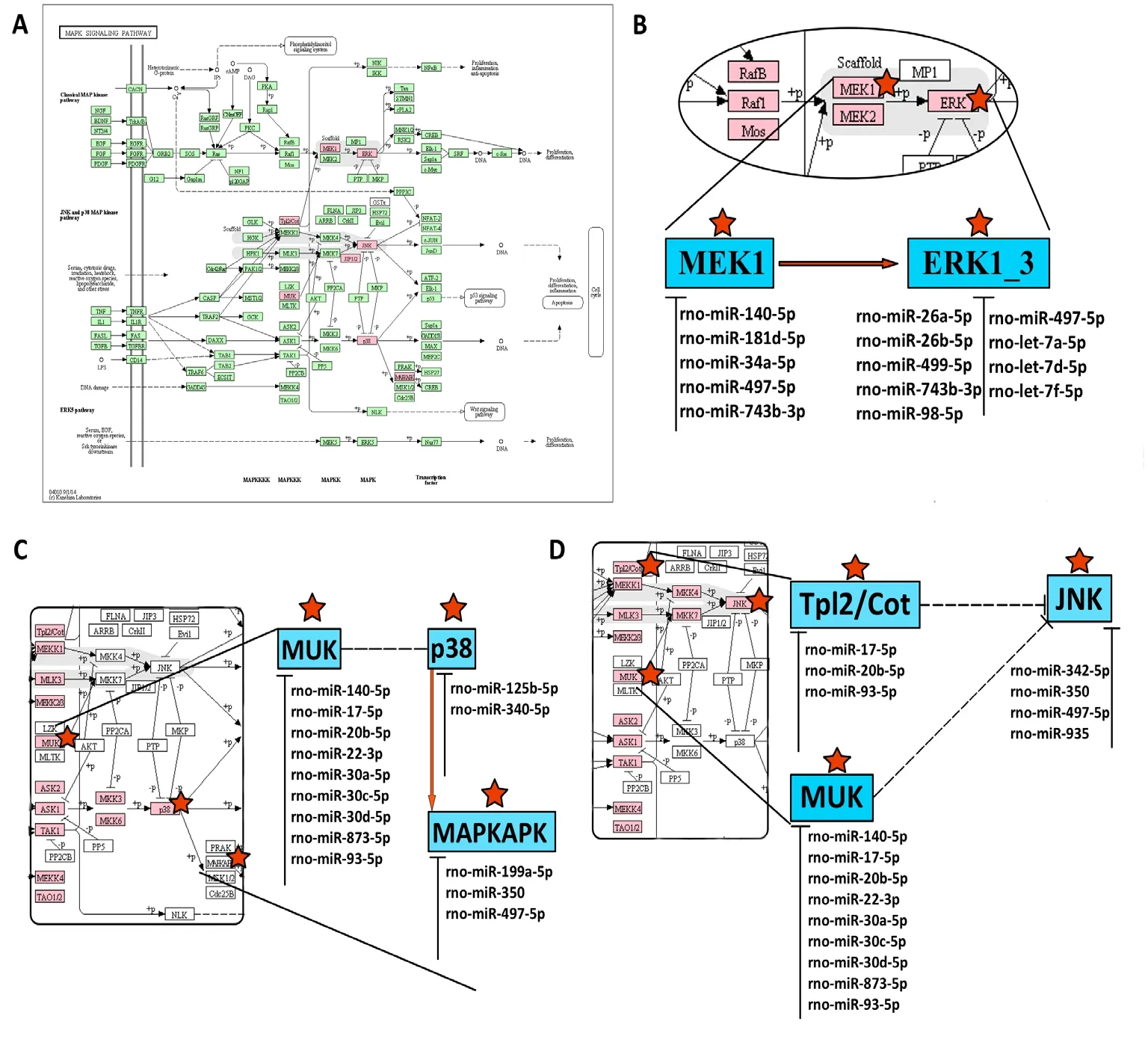
Figure 4 Potential correlations between differentially expressed miRNAs and MAPK pathways related to SCIRI.
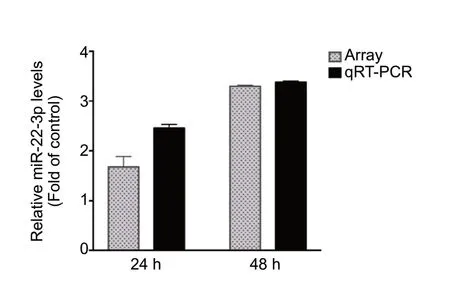
Figure 5 qRT-PCR analysis confirmed the microarray detection of key miRNAs aberrantly expressed at both 24 and 48 hours after SCIRI.
The MAPK signaling pathway reportedly plays essential roles in responding to external stimuli during ischemia-reperfusion. ERK1/2, JNK, and p38 are three major signaling cascades in the MAPK signaling transduction pathway. MAPK/ERK1/2 activates ERK1/2 and exhibits a close relationship with cell survival, differentiation, and proliferation based on the association with neurotropic factors,which are also involved in myocardial ischemia-reperfusion injury (Boulton et al., 1991; Segal and Greenberg, 1996; Jiang et al., 2013). Nuclear factor-κB is induced through the MAPK/ERK1/2 signaling pathway to regulate anti-apoptotic mechanisms in SCIRI lesion tissues (Lu et al., 2010). Furthermore, MAPK/p38/JNK is regarded as a stress-induced kinase and plays important roles in inflammatory responses(Wang et al., 2009; Abi-Hachem et al., 2010). In this study,at 24 and 48 hours after ischemia-reperfusion, most of the predicted differentially expressed miRNA target genes were mapped to the MAPK signaling pathway (14 at 24 hours and 51 at 48 hours). These findings indicate that although the molecular mechanisms of SCIRI are complicated and remain unclear, MAPK appears to be the most critical signaling pathway. And miR-22-3p may affect SCIRI process by regulating MAPK pathway.
Despite these promising findings, there are several limitations that must be considered in the interpretation of our results. First, only three repetitions were performed for each group, which may have resulted in uncontrollable errors. Second, the current study only involved a preliminary analysis of the functions of putative miRNA-TF regulatory patterns,which require further validation by functional experiments.
Nevertheless, to the best of our knowledge, this study is the first to develop a general landscape of aberrant miRNA-TF regulatory patterns in a SCIRI model. These novel findings provide important clues for future studies on SCIRI and a theoretical foundation for the development of novel therapeutic approaches.
Acknowledgments:We would like to thank the School of Public Health,Jilin University for providing the Sprague-Dawley rats and animal surgical equipment.
Author contributions:Study design: ZGL; study implementation: ZYX,YL, JHJ and HL; data analysis: ZYX and ZGL; sample collection: YL and HL; manuscript writing: ZGL; manuscript reviewing: XYY. All authors approved the final version of the paper.
Conflicts of interest:The authors declare that there are no conflicts of interest associated with this manuscript.
Financial support:This work was supported by the National Natural Science Foundation of China, No. 81350013 (to XYY). The funding source had no role in study conception and design, data analysis or interpretation, paper writing or deciding to submit this paper for publication.
Institutional review board statement:All experimental procedures were approved by the Animal Ethics Committee of Jilin University, China[approval No. 2020 (Research) 01]. The experimental procedure followed the United States National Institutes of Health Guide for the Care and Use of Laboratory Animals (NIH Publication No. 85-23, revised 1996).
Copyright license agreement:The Copyright License Agreement has been signed by all authors before publication.
Data sharing statement:Datasets analyzed during the current study are available from the corresponding author on reasonable request.
Plagiarism check:Checked twice by iThenticate.
Peer review:Externally peer reviewed.
Open access statement:This is an open access journal, and articles are distributed under the terms of the Creative Commons Attribution-Non-Commercial-ShareAlike 4.0 License, which allows others to remix, tweak, and build upon the work non-commercially, as long as appropriate credit is given and the new creations are licensed under the identical terms.
Additional files:Additional Figure 1:Putative miRNA-TF regulatory patterns in SCIRI for up-regulated miRNAs in the 48-hour group.Additional Figure 2:Putative miRNA-TF regulatory patterns in SCIRI for down-regulated miRNAs in the 48-hour group.Additional Table 1:Information for miRNA target genes.Additional Table 2:Pattern details.Additional Table 3:Transcription factors (TFs) information.
杂志排行
中国神经再生研究(英文版)的其它文章
- Muscovite nanoparticles mitigate neuropathic pain by modulating the inflammatory response and neuroglial activation in the spinal cord
- Knocking down TRPM2 expression reduces cell injury and NLRP3 inflammasome activation in PC12 cells subjected to oxygen-glucose deprivation
- Neuroprotective mechanisms of ε-viniferin in a rotenone-induced cell model of Parkinson’s disease:significance of SIRT3-mediated FOXO3 deacetylation
- Amyloid-beta peptide neurotoxicity in human neuronal cells is associated with modulation of insulin-like growth factor transport, lysosomal machinery and extracellular matrix receptor interactions
- Sequencing analysis of matrix metalloproteinase 7-induced genetic changes in Schwann cells
- Intraoperative single administration of neutrophil peptide 1 accelerates the early functional recovery of peripheral nerves after crush injury