Regulation of β2-adrenoceptors in brain glia: implications for neuroinflammatory and degenerative disorders
2020-04-29KarenM.Ryan,AndrewHarkin
Noradrenaline:Within the central nervous system (CNS), the primary source of the catecholamine neurotransmitter noradrenaline is the locus coeruleus (LC) in the pontine tegmentum, with LC neurons projecting to almost all regions of the brain and spinal cord. Following its release from LC neurons, noradrenaline has wide ranging effects. For example, noradrenaline is the endogenous agonist for G-coupled α- and β-adrenoceptors that are expressed on many cell types, including neurons and glia, in both the peripheral nervous system and CNS. It is via these receptors that noradrenaline exerts its anti-inflammatory and neurotrophic effects in the brain. Noradrenaline additionally has adrenoceptor-independent neuroprotective actions, and as such plays a role in free radical scavenging and reducing oxidative stress (Feinstein et al., 2016).
Noradrenaline and neurodegenerative disease:Owing to the numerous functions of noradrenaline, dysregulation of the LC and noradrenergic signaling can have broad ranging effects and has been suggested to contribute to neurodegenerative diseases such as Alzheimer’s disease, Parkinson’s disease, and multiple sclerosis (MS) (Feinstein et al., 2016). These conditions are generally characterized by an unresolved inflammatory response with chronic activation of glia(Marien et al., 2004) and sustained production of pro-inflammatory and potentially neurotoxic cytokines. Notably, studies have shown that noradrenaline can suppress the proliferation of microglia and limit the expression of inflammatory markers in gliain vitro(Feinstein et al., 2016).In vivo, depletion of noradrenaline increases the inflammatory response to the pathogenic protein amyloid-β1-42and exacerbates the severity of experimental autoimmune encephalomyelitis, an animal analog of MS, while increasing noradrenergic tone has been shown to attenuate inflammation in these disease models. Drugs that target the noradrenergic system to enhance extrasynaptic concentrations of the transmitter are neuroprotective in animal models of neurodegenerative disease, with effects mediated by suppression of the expression of inflammatory cytokines, chemokines, and cell adhesion molecules following central and systemic inflammatory challenge, in addition to improving functional recovery after ischemia, and promoting neuronal survivalin vivo(O’Neill and Harkin, 2018).
Noradrenaline and glial adrenoceptors:It is now widely accepted that within the CNS the neuroprotective effects of noradrenaline are primarily orchestrated by its endogenous anti-inflammatory and neurotrophic properties, mediated predominantly through its actions at glial β2-adrenoceptors (Figure 1A). Direct stimulation of glial β2-adrenoceptorsin vitrowith noradrenaline or β2-adrenoceptor agonists suppresses nuclear factor kappa-light-chain-enhancer of activated B cells (NFκB), induces the expression of anti-inflammatory mediators and negative regulators of the interleukin (IL)-1 system (IL-1RA and IL-1RII),and protects against IL-1β-induced neurotoxicity, while stimulation of central β2-adrenoceptorsin vivoincreases the expression of the broad spectrum anti-inflammatory cytokine IL-10 and its downstream mediator Suppressor of cytokine signaling 3 (O’Neill and Harkin, 2018). Moreover, stimulation of β2-adrenoceptors induces neurotrophin expression, including brain derived neurotrophic factor, nerve growth factor beta, glial derived neurotrophic factor (GDNF), and basic fibroblast growth factorin vitroandin vivo(Culmsee et al., 1999; Day et al.,2014), and induces neurite outgrowth (Day et al., 2014). The increase in GDNF following β2-adrenoceptor stimulation may be of particular importance from a neuroprotection standpoint since central infusion of GDNF can prevent dopaminergic neurodegeneration in a rat model of Parkinson’s disease and increase dopamine storage and improve motor function in patients with this disease.Thus, noradrenaline has been proposed to have a bi-modal neuroprotective role through its effects on the downregulation of microglial pro-inflammatory mediator expression and the enhancement of astrocytic growth factor production and the promotion of neurotrophic effects (O’ Neill and Harkin, 2018).
It has been proposed that the demyelination associated with the progression of MS may be due to a lack of β2-adrenoceptors on astrocytes since it has been reported that β2-adrenoceptors are absent from astrocytes in both normal appearing white matter and astrogliotic plaques in the white matter of patients with MS, though no association has been found between polymorphisms of the β2-adrenoceptor gene and the incidence of MS (De Keyser et al., 2004). Such a phenomenon suggests that endogenous noradrenaline may fail to elicit anti-inflammatory effects in the CNS, thus contributing to the dysregulation of inflammatory markers characteristically observed in MS.
Regulation of the β2-adrenoceptor by inflammatory stimuli:We recently investigated the effects of an immune stimulus comprising bacterial endotoxin lipopolysaccharide (LPS) and interferon-gamma (IFN-γ) on β2-adrenoceptors in mixed and enriched astrocytic and microglial cultures in a bid to determine if induction of inflammation could contribute to a loss of the receptor. Previous studies have shown that inflammation can reduce β-adrenoceptor expression and responsiveness in the respiratory system, including tracheal smooth muscle, lung tissue, and airway smooth muscle cells (Ryan et al., 2019). Primary rat mixed glial cells were treated with LPS and IFN-γ to mimic the neuroinflammatory environment of MS, since LPS drives expression of the pro-inflammatory cytokines IL-1β and tumor necrosis factor-α and combined with the T-cell (Th1) cytokine IFN-γ is representative of the inflammatory milieu reported in MS brain lesions.Glial β2-adrenoceptors were downregulated at both the mRNA and protein levels following exposure of mixed glial cultures (70% astrocytes, 30% microglia) to LPS + IFN-γ (Figure 1B; Ryan et al., 2019). Furthermore, exposure of mixed glia to LPS + IFN-γ decreased β2-agonist-stimulated production of the intracellular second messenger cyclic adenosine monophosphate (cAMP). Notably, LPS +IFN-γ did not impact on intracellular cAMP accumulation in response to forskolin, a direct activator of adenylate cyclase. Thus, the reduction in cAMP induced by LPS + IFN-γ is an event specific to the β2-adrenoceptor itself and not due to an effect on intracellular signaling where regulation of the β2-adrenoceptor is independent of effects on adenylate cyclase. Moreover, the effects of LPS + IFN-γ on reducing expression appear specific to the β2-adrenoceptor and do not generalize to other G-coupled receptors such as the adenosine A2Areceptor or the β1-adrenoceptor, expression of which were increased by LPS + IFN-γ. Pre-treatment with the glucocorticoid dexamethasone, a drug commonly used in the treatment of MS, suppressed the LPS + IFN-γ-induced inflammatory response and blocked the decrease in β2-adrenoceptor mRNA (Figure 1B).
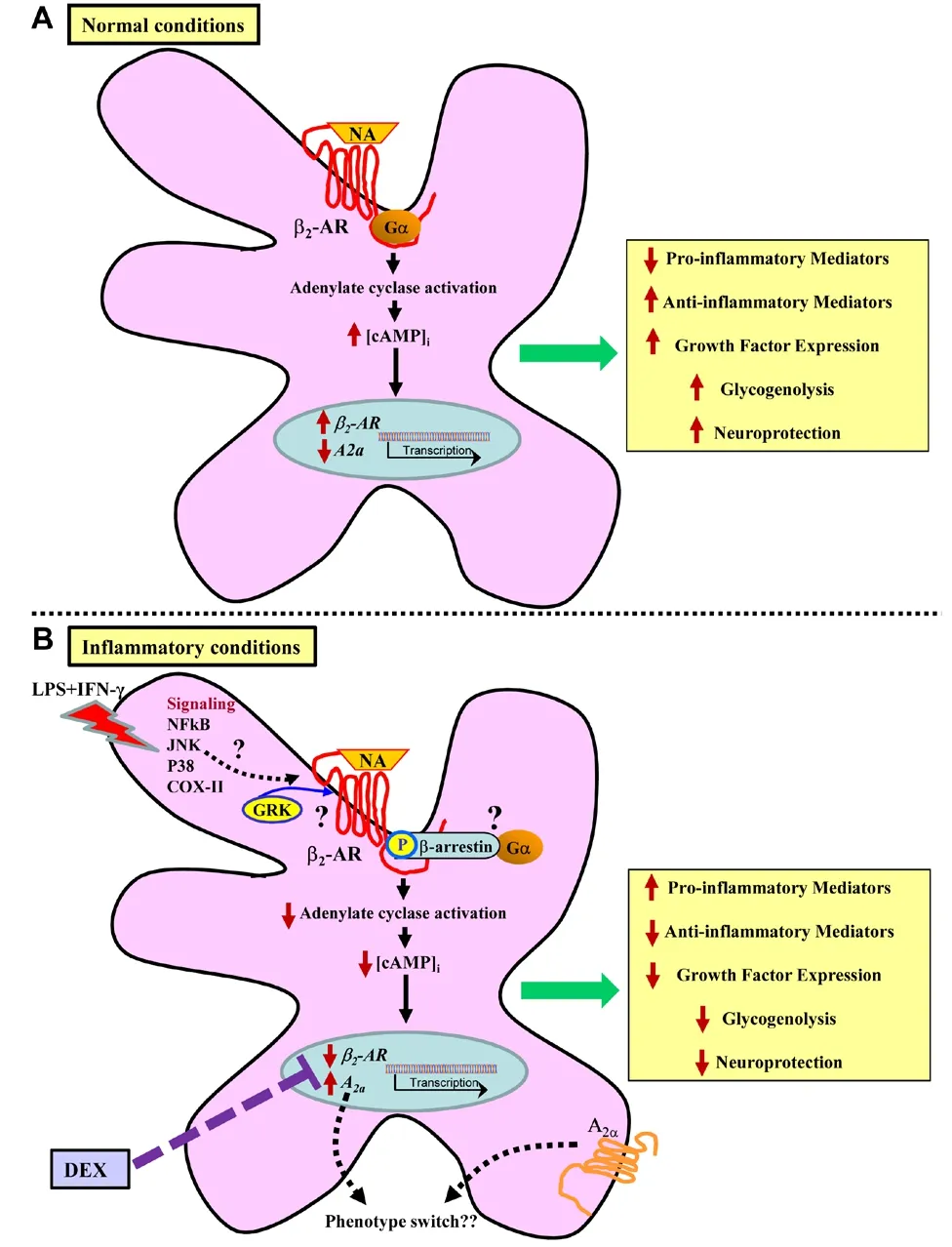
Figure 1 Glial β2-adrenoceptor function in normal and inflammatory conditions.
Mechanisms of inflammatory associated β2-adrenoceptor downregulation:The decrease in receptor function observed in mixed glial cultures following LPS+ IFN-γ exposure is suggestive of receptor internalization, decreased translation of mRNA to receptor protein, or both. The simplest explanation for this is that a decline in transcription would lead to diminished translation of β2-adrenoceptor protein, and this in turn would lead to reduced receptor cell surface expression and function as observed. However, treatment with dexamethasone restores β2-adrenoceptor mRNA levels but fails to restore function of the receptor. It is possible that the time period required for dexamethasone to potentially restore the receptor to the cell surface and for it to become functional is longer than the time required to revert its effects on receptor mRNA levels. However, it may also be the case that the inflammatory-induced decrease in β2-adrenoceptor mRNA and function are mediated by different mechanisms. Thus, there are several other potential explanations for reduced cell surface expression and function of the β2-adrenoceptor (Figure 1B). One such possibility is that LPS + IFN-γ increases the recruitment of G-protein receptor kinases and β-arrestin to the receptor,which are responsible for the phosphorylation of and subsequent internalization and desensitization of the β2-adrenoceptor (Pitcher et al., 1999; Luttrell and Lefkowitz, 2002), respectively. Another possibility is that signal transduction pathways activated by LPS play a role. While the effects of pharmacological inhibitors of p38, c-Jun N-terminal kinase, NFκB, and cyclooxygenase-2 on β2-adrenoceptor expression were assessed individually, to no effect (Ryan et al., 2019),it may be the case that a combination of these signaling pathways is required to mediate downregulation of the receptor. microRNAs, small non-coding RNAs that function in post-transcriptional regulation, have also been demonstrated to mediate β2-adrenoceptor regulation (Wang et al., 2011); thus, their role in inflammation-induced β2-adrenoceptor downregulation warrants further investigation. Overall, the precise mechanisms leading to a decrease in β2-adrenoceptor mRNA and cell surface expression in mixed glial cultures following exposure to an inflammatory stimulus remain to be fully elucidated. Moreover, it remains to be determined whether incubation of cells with LPS + IFN-γ leads to a reduced ability of noradrenaline or β2-agonists to dampen the inflammatory response in glia and if this can be blocked by dexamethasone.
Implications of inflammatory associated β2-adrenoceptor downregulation:An inflammatory-related decrease in β2-adrenoceptor expression and function could result in an inability of endogenous noradrenaline to elicit anti-inflammatory and neuroprotective effects within the CNS. As such, downregulation of the receptor may contribute to neurodegeneration owing to a number of consequences. First, the transcription of trophic factors in astrocytes is dependent on cAMP signaling, mostly via the β2-adrenoceptor. Hence, loss of the receptor and the associated decrease in cAMP would likely result in a loss of neurotrophic support.Second, production of pro-inflammatory molecules could become dysregulated in the absence of noradrenergic actions at β2-adrenoceptors leading to damage of neurons and oligodendrocytes. Third, most of the brain’s energy source is stored in astrocytes in the form of glycogen. Glycogenolysis is dependent on stimulation of astrocytic β2-adrenoceptors and cAMP production; therefore, decreased production of intracellular cAMP would lead to reduced glycogenolysis and further impact on energy supply to glia and neurons in the CNS. Excessive Ca2+influx due to failure of the Na+/K+pump, which relies on ATP to function, and increased activity of Ca2+-dependent degradative enzymes, such as proteases and phospholipases, are liable to induce neuronal damage.
A decrease in β2-adrenoceptor expression may also result in a phenotypic switch of glia. It has been shown that under resting conditions microglia express β2-adrenoceptor mRNA, though following exposure to an inflammatory stimulus β2-adrenoceptor expression is reduced and there is a switch towards expression of α2A-adrenoceptors. Notably, noradrenaline mediates microglial process retraction and modulates microglial motility under resting and activation states via β2-adrenoceptors and α2A-adrenoceptors, respectively. Moreover, upregulation of the adenosine A2Areceptor can result in a switch in the chemotactic activity of microglial cells from being attracted to ATP released at a site of injury to assuming an amoeboid shape and being repelled from ATP in the presence of LPS. This switch ultimately results in decreased phagocytic ability of microglia.LPS + IFN-γ increased adenosine A2Areceptor expression (Figure 1B; Ryan et al., 2019). In certain neurodegenerative conditions, notably Alzheimer’s disease,adenosine A2Areceptors are upregulated in neurons and astrocytes. A2Aoverexpression promotes transcriptional changes affecting inflammatory markers,including IL-1β and other genes related to immune response, angiogenesis, and cell activation, in primary astrocytic cultures (Paiva et al., 2019).
It is tempting to speculate that suppression of inflammatory associated β2-adrenoceptor downregulation by dexamethasone, a drug commonly used in the treatment of MS, may contribute to its therapeutic efficacy. Downregulation of glial β2-adrenoceptors, following exposure to inflammatory stimuli as described,may ultimately be detrimental in the CNS given that the anti-inflammatory action of endogenous noradrenaline may be impaired.
In vivo context:The β2-mediated effects of noradrenaline clearly represent a useful therapeutic strategy for the treatment of neuroinflammatory and neurodegenerative disease. Pharmacological targeting of β2-adrenoceptors has been shown to be neuroprotective following acute exposure to LPS (4 hours) in the inflammatory rat model of Parkinson’s disease (O’Neill et al., 2019) and treatment with the noradrenaline re-uptake inhibitor atomoxetine alone and in combination with the α2-adrenoceptor antagonist idazoxan attenuates loss of dopamine and associated motor deficits in the acute (4 hours) LPS inflammatory rat model of Parkinson’s disease (Yssel et al., 2018). However, it remains to be determined if prolongedin vivoexposure to LPS, for example 24 hours to replicate ourin vitromodel or longer, results in the downregulation of either β2-adrenoceptor expression or function in the brain. If this phenomenon identifiedin vitroalso occurs in thein vivocontext then it will be of importance to elucidate strategies to prevent β2-adrenoceptor downregulation in order to preserve the endogenous anti-inflammatory and neuroprotective actions of noradrenaline. Moreover,additional studies are required to determine if noradrenaline or noradrenergic agonists can combat neuroinflammatory or neurodegenerative processes in conditions of chronic inflammation.
Conclusion:Further work is required to fully elucidate the mechanisms involved in inflammation-induced glial β2-adrenoceptor downregulation. Better understanding of glial β2-adrenoceptor regulation may be of value for the development of treatments for neuroinflammatory disorders such as MS and chronic neurodegenerative conditions including Parkinson’s and Alzheimer’s diseases.
Karen M. Ryan, Andrew Harkin*
Trinity College Institute of Neuroscience, Trinity College Dublin; Department of Psychiatry, St. Patrick’s University Hospital, Trinity College Dublin, Dublin,Ireland (Ryan KM)
Neuropsychopharmacology Research Group, School of Pharmacy and Pharmaceutical Sciences & Trinity College Institute of Neuroscience, Trinity College Dublin, Dublin, Ireland (Harkin A)
*Correspondence to:Andrew Harkin, PhD, aharkin@tcd.ie.
orcid:0000-0001-9734-216X (Andrew Harkin)
Received:January 16, 2020
Peer review started:January 21, 2020
Accepted:March 4, 2020
Published online:May 11, 2020
doi:10.4103/1673-5374.282255
Copyright license agreement:The Copyright License Agreement has been signed by both authors before publication.
Plagiarism check:Checked twice by iThenticate.
Peer review:Externally peer reviewed.
Open access statement:This is an open access journal, and articles are distributed under the terms of the Creative Commons Attribution-NonCommercial-Share-Alike 4.0 License, which allows others to remix, tweak, and build upon the work non-commercially, as long as appropriate credit is given and the new creations are licensed under the identical terms.
Open peer reviewer:Melissa Schepers, Hasselt University, Belgium.
Additional file:Open peer review report 1.
杂志排行

中国神经再生研究(英文版)的其它文章
- The role of the TrkB-T1 receptor in the neurotrophin-4/5 antagonism of brain-derived neurotrophic factor on corticostriatal synaptic transmission
- Could non-invasive brain-stimulation prevent neuronal degeneration upon ion channel re-distribution and ion accumulation after demyelination?
- The role of exercise in brain DNA damage
- Combined effect of repetitive transcranial magnetic stimulation and physical exercise on cortical plasticity
- Should mast cells be considered therapeutic targets in multiple sclerosis?
- Neuroprotection mediated by natural products and their chemical derivatives