阿齐沙坦片体外溶出方法
2020-04-27杨锐李玲玲李婷丛海建姜锋
杨锐 李玲玲 李婷 丛海建 姜锋
摘 要 目的:优化阿齐沙坦片体外溶出条件,为该品种的仿制药一致性评价工作提供基础。方法:采用中国药典2015年版四部0931第二法(桨法),pH 1.0(含0.03% SDS)、pH 4.5(含0.4% SDS)、pH 6.8、水(含0.2% SDS)为溶出介质,转速为50 r/min,HPLC 法测定溶出曲线。结果:阿齐沙坦在4种溶出介质中专属性良好,平均回收率分别为99.01%(RSD 1.31%)、99.57%(RSD 1.04%)、99.87%(RSD 1.62%)、101.67%(RSD 1.53%)。结论:建立的溶出曲线测定方法准确、可靠,可为阿齐沙坦片的仿制药体外溶出评价提供参考。
关键词 阿齐沙坦片 溶出曲线 体外溶出
中图分类号:R972.4; R917 文献标志码:A 文章编号:1006-1533(2020)07-0065-06
Study on in vitro dissolution method of azilsartan tablets
YANG Rui*, LI Lingling, LI Ting, CONG Haijian, JIANG Feng**
(Shanghai Haini Pharmaceutical Co., Ltd., Yangtze River Pharmaceutical Group, Shanghai 201318, China)
ABSTRACT Objective: To optimize the in vitro dissolution conditions of azilsartan tablets and provide experimental basis for generic drugs quality consistency evaluation. Methods: According to the second dissolution method (paddle method) stated in 0931 of Chinese Pharmacopeia (2015 Edition), the dissolution of azilsartan in different media (pH 1.0, containing 0.03% SDS; pH 4.5, containing 0.4% SDS; pH 6.8; water, containing 0.2% SDS) with rotation of 50 rpm was determined by HPLC. Results: Azilsartan tablets had good specificity in the four kinds of dissolution media and their average recoveries were 99.01% (RSD 1.31%), 99.57% (RSD 1.04%), 98.77% (RSD 1.62%) and 101.67% (RSD 1.53%), respectively. Conclusion: The established method for the determination of azilsartan dissolution is accurate and reliable and can provide a reference for in vitro dissolution evaluation of azilsartan tablets.
KEy WORDS azilsartan tablet; dissolution profile; in vitro dissolution
阿齊沙坦是新一代选择性AT1亚型血管紧张素Ⅱ受体拮抗剂(ARBs)类抗高血压药,与血管紧张素转化酶抑制剂类抗高血压药比,具有不会引起干咳、平稳降压的优点。2012年1月,日本武田药品工业株式会社开发的阿齐沙坦片(商品名:Azilva)在日本获批上市[1];2011年2月25日美国FDA也批准其前药阿齐沙坦酯片(商品名:Edarbi)用于治疗成人高血压;2014年日本MHLW研究证明阿齐沙坦片单独或联合用药均具有平稳持久降血压作用,而且还能通过部分激活过氧化物酶体增殖物激活受体γ而对糖尿病患者产生潜在的保护作用。临床研究显示,阿齐沙坦较奥美沙坦与坎地沙坦降压作用更强,更持久,降压效果更稳定。目前,阿齐沙坦片尚未在中国上市。
药物溶出速率决定其在体内的吸收和利用程度,提高溶出速度和程度是改善其生物利用度的有效途径,进而提高药物的疗效[1-2]。阿齐沙坦为难溶性药物,属于Biopharmaceutics Classification System(BCS)Ⅱ类药物,其原研片在pH 1.0盐酸溶液、水、pH 4.5醋酸盐缓冲液中不能完全溶出,而在pH 6.8磷酸盐缓冲液中溶出速度较快,若以pH 6.8磷酸盐缓冲液作为溶出介质与原研片进行溶出曲线比较,势必缺乏较好的曲分力[3]。本研究通过测定阿齐沙坦原料药在不同溶出介质中的饱和溶解度,利用原研片完成提高该药溶出度条件的初步筛选与优化,并验证了溶出曲线测定方法,取得具有区分力的溶出曲线,从而可在研发阶段更好地评价阿齐沙坦片的体外溶出行为。
1 材料和方法
1.1 试药
阿齐沙坦原料药(批号16033101-1)、阿齐沙坦对照品(批号16052401,纯度99.71%)和阿齐沙坦自制片(批号:17020703)均来自扬子江药业集团上海海尼药业有限公司;阿齐沙坦原研片(日本武田制药,批号AH332、AH382,规格20 mg)。
1.2 试剂
十二烷基硫酸钠(SDS,批号20160110,分析纯,天津科密欧化学试剂有限公司);磷酸(批号150310,色谱纯,Fisher公司);甲醇(批号I830907617,色谱纯,Merck公司);其他化学试剂均为市售分析纯。
1.3 仪器
1260高效液相色谱仪(Agilent公司);U3000高效液相色谱仪(Thermo公司);AB204-S电子天平(Mettler Toledo公司);THZ-82水浴恒温振荡器(金坛市杰瑞尔电器有限公司);RC807D溶出仪(天大天发公司)。
1.4 方法
1.4.1 色谱条件
以十八烷基硅烷键合硅胶为填充剂(Welch Ultimate XB-C18,4.6 mm×250 mm,5 mm),以0.5%磷酸水溶液-甲醇(30∶70)为流动相,检测波长为251 nm,柱温为30 ℃,流速为1.2 ml/min,进样体积为10 μl;理论板数按阿齐沙坦峰计算,应不低于3 500。
1.4.2 pH-溶解度曲线测定
取阿齐沙坦原料药50~70 mg,置8支具塞西林瓶中,分别精密移取pH 1.0、3.8、4.5、5.5、6.0、6.8、7.6、8.0的盐酸溶液及缓冲盐溶液[4]10 ml,平行两份,置37 ℃水浴中振荡过夜,使之形成过饱和溶液,滤过,取续滤液经HPLC法测定。
1.4.3 阿齐沙坦在不同溶出介质中的饱和溶解度测定
取阿齐沙坦原料药50~70 mg,置具塞西林瓶中,分别精密移取不同SDS浓度的pH 1.0盐酸溶液、水、pH 4.5醋酸盐缓冲液各10 ml,平行三份,置37 ℃水浴振荡24 h,使之形成过饱和溶液,静置,精密移取上清液1 ml于具塞试管中,精密加入70%甲醇溶液8 ml,摇匀,即得供试品溶液,经HPLC法测定。
1.4.4 阿齐沙坦片溶出曲线优化
分别取不同SDS浓度的pH 1.0盐酸溶液、水、pH 4.5醋酸盐缓冲液各900 ml,置溶出杯中,温度为37 ℃,桨法,转速为50 r/min;平行测定阿齐沙坦原研片6片,分别于5、10、15、30、45、60 min各时间点取溶液8 ml,不补液,用0.45 mm的滤膜过滤,弃去5 ml,取续滤液作为供试品溶液。精密称取阿齐沙坦对照品约19 mg,置20 ml量瓶中,加入甲醇溶解并稀释至刻度,摇匀,作为对照品储备液。精密量取1 ml,置50 ml量瓶中,加溶出介质稀释至刻度,摇匀,即得对照品溶液。然后,进行HPLC法测定,并计算累积溶出度。
1.4.5 阿齐沙坦片溶出曲线方法的建立
1)供试品溶液制备 取阿齐沙坦片,照溶出度测定法(《中国药典》2015年版四部通则0931第二法桨法)测定,分别取含0.03% SDS的pH 1.0盐酸溶液(简称pH 1.0)、含0.4% SDS的pH 4.5醋酸盐缓冲液(简称pH 4.5)、pH 6.8磷酸盐缓冲液(简称pH 6.8)、含0.2% SDS水(水)为溶出介质,介质体积900 ml,转速50 r/min,温度(37±0.5)℃,分别于5、10、15、30、45、60 min取溶液8 ml,不补液,用0.45 mm的滤膜过滤,弃去5 ml,取续滤液作为供试品溶液。
2)对照品溶液的制备 取阿齐沙坦对照品约19 mg,精密称定,置20 ml量瓶中,加甲醇溶解并稀释至刻度,摇匀,作为对照品储备液。精密量取1 ml,置50 ml量瓶中,加各溶出介质稀释至刻度,摇匀,作为对照品溶液。
1.4.6 方法学研究
1)专属性 取阿齐沙坦片空白辅料、阿齐沙坦自制片及阿齐沙坦对照品,制备供试品和对照品溶液。分别精密量取上述各溶液10 μl,进行色谱分析。
2)滤膜吸附 分別于15 min(pH 6.8磷酸盐缓冲液)、30 min(含0.2% SDS的纯化水、含0.03%SDS的pH 1.0盐酸溶液)、45 min(含0.4%SDS的pH 4.5醋酸盐缓冲液)时,取各溶出介质溶出液各10 ml,离心,取上清液作为供试品控制溶液;另取上述各溶出介质溶出液10 ml,经滤膜滤过,取初滤液及分别弃去初滤液0、2、4、5、6 ml后取续滤液,作为供试品溶液。分别精密量取上述各溶液10 μl,进行色谱分析。
3)准确度 按照溶出度(以85%计)的40%、100%、120%设计回收率试验。精密称取阿齐沙坦对照品制备成对照品溶液;按处方比例称取空白辅料9份,分别加入上述对照品溶液2、5、6 ml,各制备3份,用相应溶出介质稀释至刻度,摇匀,分别于(37±0.5)℃下振摇30 min后取样,过滤(弃去初滤液5 ml),取续滤液作为供试品溶液。然后进行色谱分析。
4)溶液稳定性 将对照品溶液和供试品溶液分别在常温密封条件下放置0、1、2、4、6、8、10、12、18、24、30、36、38 h。分别取样进行色谱分析。
2 结果
2.1 阿齐沙坦溶解度与阿齐沙坦原研片溶出曲线
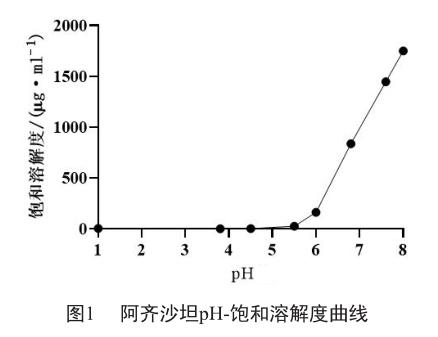
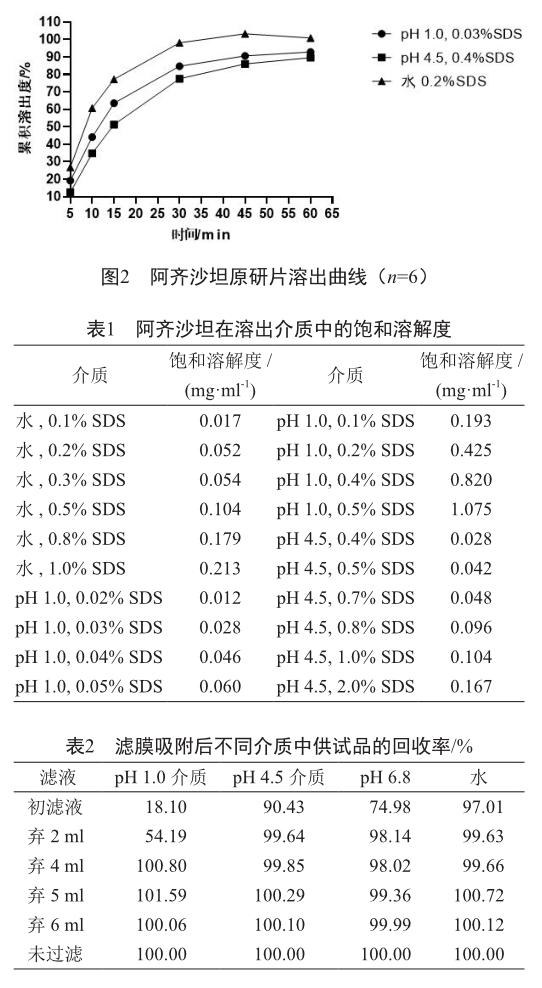
阿齐沙坦在酸性pH条件下难溶,而在碱性pH条件下有较高的溶解度(图1);不同溶出介质中加入适宜浓度的SDS可增加其溶解度(表1);经对在不同溶出介质中具有区分力的表面活性剂浓度的筛选和优化[3],得到阿齐沙坦原研片在30~45 min时的累积溶出度≥85%,达到溶出平台(图2)。
2.2 方法学研究
2.2.1 专属性
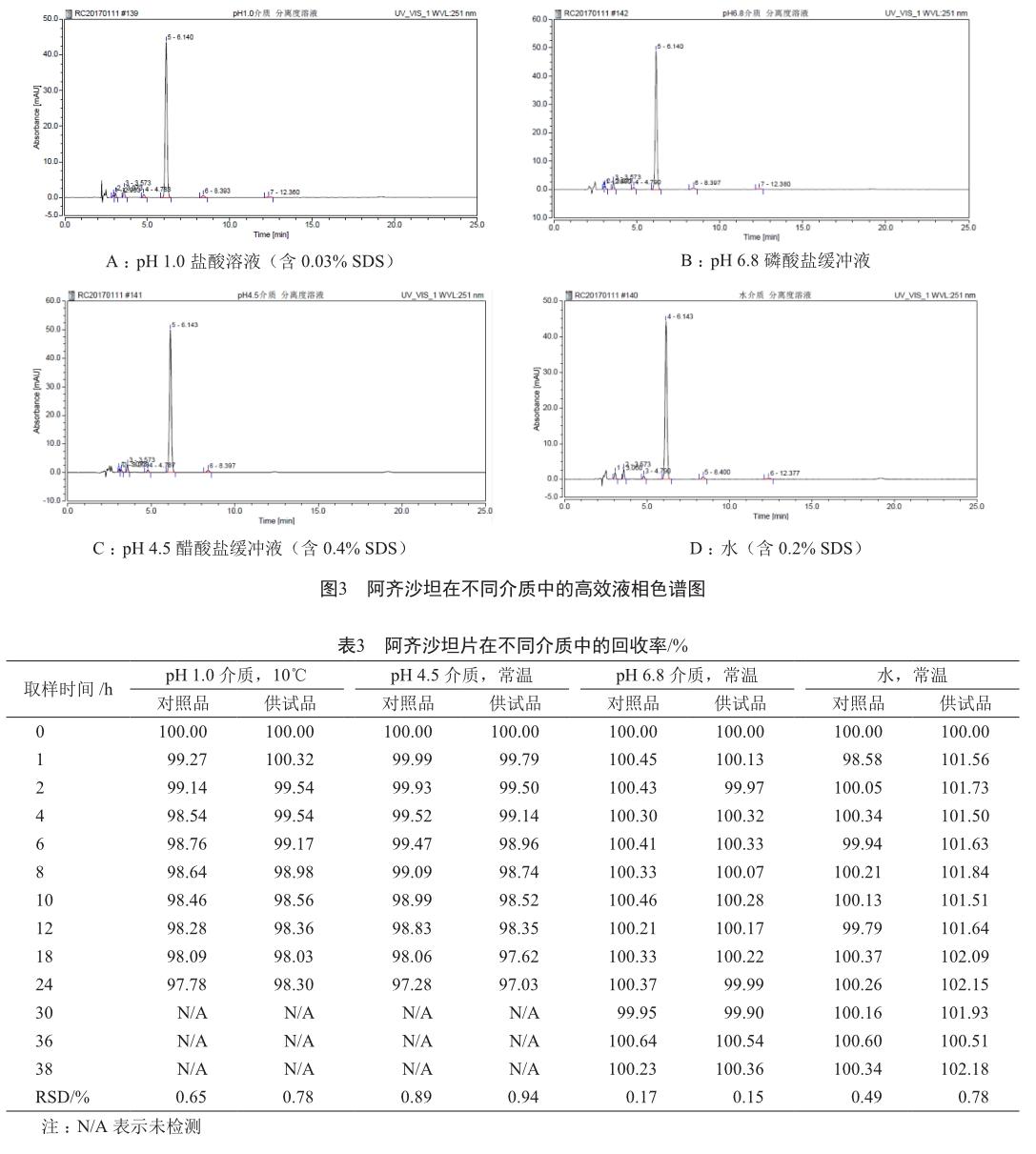
阿齐沙坦在各介质溶液中的分离度均远大于1.5,达到完全分离(图3)。四种溶出介质和空白辅料对本品溶出度测定没有干扰,表明其在该色谱条件下的专属性良好。
2.2.2 滤膜吸附
供试品溶液均能保证滤膜吸附率小于1.0%(回收率大于99.0%)(表2)。
2.2.3 准确度
阿齐沙坦在pH 1.0盐酸溶液(含0.03% SDS)、pH 4.5醋酸盐缓冲液(含0.4% SDS)、pH 6.8磷酸盐缓冲液、水(含0.2% SDS)中的平均回收分别为99.01%、99.57%、99.87%、101.67%,RSD(n=9)依次为1.31%、 1.04%、1.62%、1.53%,表明阿齐沙坦在四种溶出介质中回收率良好。
2.2.4 溶液稳定性
在溶液稳定时间范围内,不同时间点供试品溶液和对照品溶液峰面积与0时相应溶液峰面积对比在98%~102%范围内。在pH 1.0和pH 4.5介质中样品稳定时间为12 h、而在pH 6.8介质和水介质中稳定时间可达到38 h(表3)。
2.3 原研片溶出曲线测定
原研片在四种溶出介质中的平均溶出曲线见图4(n=12),表明在我们建立的溶出条件下原研片在30~45 min时的累积溶出度均可达85%以上,符合有关规定。
2.4 阿齐沙坦自制片与原研片体外溶出对比
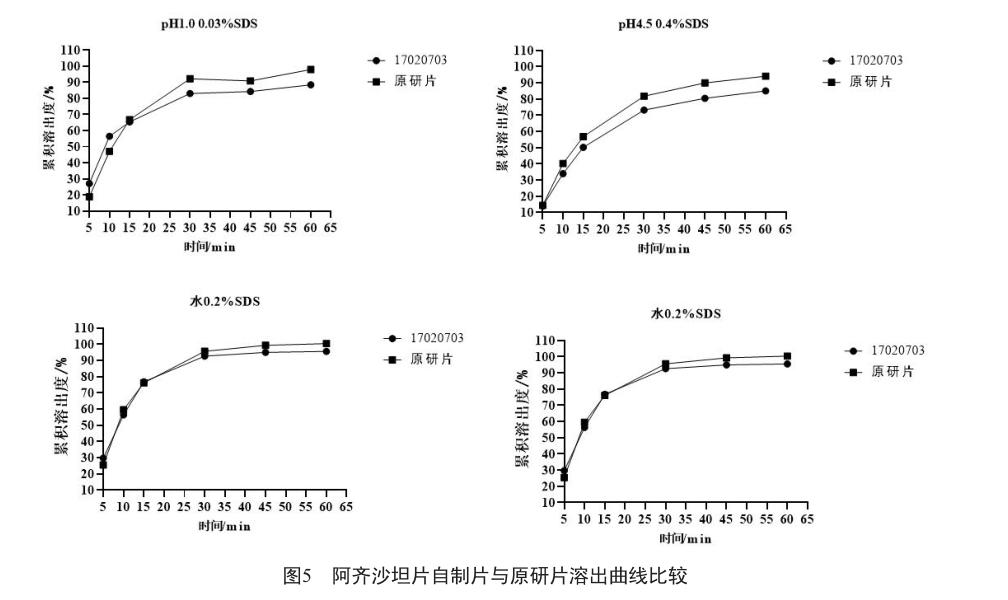
利用以上筛选的溶出条件进行自制片与原研片的体外溶出研究,自制片与原研片在4种不同介质中的溶出曲线相似(图5),体外溶出一致(f2>50)。
3 讨论
国内相关文献主要针对阿齐沙坦片溶出度进行了相关研究。如朱亚东等[5]和李瑛等[6]对阿齐沙坦片溶出度方法学进行研究,但未对提高溶出度进行方法研究;唐了平等[7]对比自制制剂与原研制剂体外溶出行为,但比较的仅为常规的pH 1.0、pH 4.5、pH 6.8和水等溶出介质,且除pH 6.8外,阿齐沙坦片在其他溶出介质中的最终累积溶出度最高仅为35.8%,溶出很少,这在研发阶段不利于评价自制制剂与原研制剂的体外溶出是否一致,对于处方工艺的筛选无指导价值;郭永斌[8]的研究虽然阐述了溶出曲线用于研究的重要性,但未对各个溶出介质的条件进行优化。本研究通过对阿齐沙坦片体外溶出条件的优化,筛选出具有区分力的溶出条件,从而能更好地指导自制制剂的研发。
阿齐沙坦为难溶性药物,阿齐沙坦片体外溶出方法的建立难点在于表面活性剂种类的选择。通过筛选阿齐沙坦片溶出介质中表面活性剂SDS的加入量,最终确定在水、pH 1.0盐酸溶液及pH 4.5醋酸盐缓冲液中的加入浓度分别为0.2%(w/v)、0.03%(w/v)、0.4%(w/v),可使阿齐沙坦片在30~45 min的累积溶出度达85%以上,从而获得具有区分力的溶出条件[3],更有利于评价阿齐沙坦片的溶出行为是否与原研制剂一致。
溶出曲线既是评价口服固体制剂仿制药体外一致性的主要技术手段,也对保证药品批间质量的一致性起着重要作用,本研究对阿齐沙坦片仿制药质量一致性评价工作起到了一定的推进作用。
参考文献
[1] 武田药品工业株式会社. 药物组合物: 中国200780037942.5[P]. 2009-09-09.
[2] 郑忠辉, 张代铭, 吴辉, 等. 阿齐沙坦的合成工艺研究[J].化工时刊, 2015, 29(8): 10-13.
[3] 谢沐风. 具有区分力的溶出曲线[J]. 中国医药工业杂志, 2014, 45(7): 687-689.
[4] 国家药典委员会. 中华人民共和国药典2015年版四部[M]. 北京: 中国医药科技出版社, 2015: 通则8004.
[5] 朱亚东, 黄柳. 阿齐沙坦片溶出度方法学研究[J]. 化工设计通讯, 2018, 44(8): 197-199.
[6] 李瑛, 张腾, 戴根来. HPLC法测定阿齐沙坦片的溶出度[J]. 安徽医药, 2015, 19(10): 1887-1888.
[7] 唐了平, 产运霞, 马贵红, 等. 阿齐沙坦片國产品溶出度试验方法的建立及与原研品体外溶出行为比较[J]. 中国药房, 2014, 25(17): 1609-1611.
[8] 郭永斌. 阿齐沙坦片的制备及质量研究[D]. 石家庄: 河北医科大学, 2016.
