基于分子对接技术虚拟筛选天然产物抗MRSA活性成分*
2020-04-19武红莉
雷 蕾,杨 乐,亢 力,武红莉,王 忠**
(1. 中国中医科学院中医药信息研究所 北京 100700;2. 中国中医科学院中医临床基础医学研究所 北京 100700)
耐甲氧西林金黄色葡萄球菌(methicillin-resistant staphy lococcus aureus,MRSA)是多药耐药性感染的最常见原因之一,具有显着的发病率和死亡率,已成为医院感染和社区获得性感染的重要病原菌之一。多数的MRSA 感染会引起菌血症,心内膜炎和肺炎等疾病,危及生命[1]。目前,MRSA 作为一种超级细菌,仅对万古霉素、替考拉宁、利奈唑胺等少数抗菌药物敏感。然而,万古霉素有较强的肾毒性[2],限制了其在临床的应用,而且耐万古霉素金黄色葡萄球菌(hVRSA)已在全球检测。临床研究表明,hVRSA 与MRSA 密切相关,在某些国家50%的临床MRSA 检测到hVRSA 表型[3]。因此,科学家一直在寻找新的方法来对抗MRSA抗生素耐药性的影响。
MRSA 具有多重耐药性,其产生机制是PBPs 改变的结果,高度耐药性系由于原有的PBP2 与PBP3 之间产生一种新的PBP2′(即PBP2a),低、中度耐药系由于PBPs 的产量增多或与甲氧西林等的亲和力下降所致。克服抗生素耐药性的最常见方法,除了联合使用抗生素治疗以外,就是开发新药。已经有很多从天然产物中开发出新药的例子,例如,青蒿素的开发[4]和长春花碱的开发[5]。天然产物是人类的宝库,经过多年的实验研究,许多天然产物的药理作用被测定出来。本研究期望从已经报道的具有抗菌和抑菌作用的天然产物中筛选出可能具有抗MRSA 的药物。
分子对接是通过受体的特征以及受体和药物分子之间的相互作用方式来进行药物设计的方法,主要研究分子间(如配体和受体)相互作用,并预测其结合模式和亲合力的一种理论模拟方法。近年来,分子对接方法已成为计算机辅助药物研究领域的一项重要技术。现在应用中的分子对接软件涵盖了刚性对接、半柔性对接、柔性对接等各种对接方法,在能量优化方面则使用了人工神经网络、遗传算法、模拟退火、禁忌搜索、局部搜索等各种方法。在这项研究中,本课题组使用分子对接的方法对859 种具有抗菌和抑菌作用的天然产物与常见MRSA 抑制靶标青霉素结合蛋白2a(PBP2a)进行了分子对接,以期能够找到有潜力的候选物,给新药开发人员以重要参考。
1 资料与方法
1.1 平台和软件
本研究所有工作均在Microsoft Windows 2007 操作系统中完成,采用Accelrys 公司的Discovery Studio 4.5(DS 4.5)软件。参数设置除非特殊指明,均为默认值。本研究采用的是CDOCKER程序,基于CHARMm、soft-core potentials、optional grid representation 受体配体对接。它随机搜索小分子构象,采用模拟退火算法进行优化,从而对接结果更加准确。分子ADMET 计算采用DS 4.5完成。
1.2 受体的准备
PBP2a 的3D 结构(PDB ID:3ZG0,2.60 Å)从通过晶体学研究发现,PBP2a存在变构位点和活性位点,变构结合域距离dd -转肽酶活性位点60 Å。当小分子抑制剂在变构位点时,活性位点的残基构象发生变化,从而允许配体进入[6]。3ZG0 是蛋白与两个分子的Ceftaroline(头孢洛林)共结晶体,配体Ceftaroline(ID:1W8)位于变构位点,Ceftaroline 分子(ID:AI8)位于活性位点。目前,3ZG0 已经用于抗MRSA 药物的虚拟筛选[7-8]。
本研究通过Clean Protein 工具去水加氢,再加CHARMm 力场优化,定义蛋白为受体,以其原配体位置为中心,选择半径0.50 nm 范围的残基为活性残基,将其定义sphere球,此范围内的空腔为结合口袋,最后保存备用。
1.3 配体数据的组建
从TCMDB 数据库[9]中检索得到生物活性包括“抗菌”和“抑菌”的化合物共859 个。这些化合物是实验已经证明可以抗菌或者抑菌的,但是还未进行抗MRSA 研究。首先,将化合物的sdf 格式文件导入DS4.5中,Small Molecules-prepare ligand 进一步将配体产生三维结构,加氢,产生异构体等;其次,使用Linpiski 规则进行筛选;最后,使用Small Molecules-Minimize Ligands,加CHARMm 力场进行能量最小化,保存为分子对接的配体分子集。
1.4 对接方法可信性验证
含有原配体的蛋白晶体复合结构,将原配体抽离,然后按设定的参数对接回其结合口袋,计算对接后最高打分的构象与原配体结构的均方根偏差值(RMSD),一般RMSD≤0.20 nm[10]则认为该对接方法可行,说明该套参数能较好地重现此配体与受体的结合模式。
1.5 配体分子与受体靶点对接参数
将上述配体和受体导入DS 4.5,调用CDOCKER对接模块,参数均为默认值。
3ZG0 变构位点结合口袋的位置是x=9.965744,y=52.253615,z=23.244846,结 合 口 袋 的 半 径 为11.520969。
3ZG0 活性位点结合口袋的位置是x=27.901615,y=29.847026,z=88.316846,结 合 口 袋 的 半 径 为12.825363。
1.6 结果处理
对接完成后,以原配体的对接结合能(INTERACTION ENERGY)为参考,值低于原配体的说明与蛋白结合能低于原配体,予以保留,得到潜在有活性的化合物。
1.7 ADMET计算
候选药物的ADMET 分析包括血脑屏障参数(Blood Brain Barrier,BBB)、人类肠道吸收(Human Intestinal Absorption,HIA)、药物水溶性(Aqueous Solubility)、Cytochrome P450 2D6(CYP2D6)和血浆蛋白结合参数(Plasma Protein Binding,PPB)。血脑屏障阻碍大多数药物进入大脑,使有效候选药物的开发变得困难。最近的研究报道,98%的药物在临床试验中失败,正是由于不能通过血脑屏障或达到特定的靶标。CYP2D6 参与肝脏内多种底物的代谢,药物对其的抑制作用是药物-药物相互作用的主要表现。因此,CYP2D6 抑制实验是药物发现和开发过程中监管程序的一部分。血浆蛋白结合药物分子可以影响药物的效率,因为结合部分暂时被屏蔽,而只有未结合部分可以发挥药理作用。
2 结果
2.1 可行性验证结果
本研究计算RMSD 小于0.2 nm,说明对接方法、所选用的蛋白质晶体结构和参数的设定是可行的。
2.2 变构部位分子对接
本研究将原配体Ceftaroline 对接回变构部位结合口袋得到结合能为-54.8853。完成对接后,能量低于其化合物有81 个,结合能最低的10 个化合物,见表1和图1。
2.3 活性部位分子对接
本研究将原配体Ceftaroline 对接回活性部位结合口袋,得到结合能为-65.7609,能量低于其化合物有33个,结合能最低的10个分子,见表2和图1。
2.4 候选药物的ADMET分析
变构位点和活性位点均对接较好的化合物有24 个 化 合 物 ,ADMET_BBB_Level 在 low 包 括 low 以上的化合物有9 个。本研究进行了ADMET 分析,见 表 3 和 图 2。 可 以 看 出 ,Abyssinone V 和Isogancaonin C 的血脑屏障渗透水平为中等,脑-血比在0.3:1 和1:1 之间,其余的都为低,说明脑-血比低于0.3:1。 Gancaonin C,Isogancaonin C 和 Rhamnazin 的水溶性为最佳。9 个化合物预测均无Cytochrome P450 2D6 毒性,同时都具有良好的人类肠道吸收率。Glycycoumarin 和Kuwanon A 血浆蛋白结合较好。
2.5 作用模式分析
为了研究突变体结构与活性化合物的结合亲和力,进行了作用模式的分析。结果表明,在表3 中,除了 Glycycoumarin 和 Quercetin-3-methyl ether 外,其余分子在3ZG0 变构位点上都与146 位蛋白通过范德华力或者氢键结合,而在3ZG0 活性位点上,除了范德华力和氢键外,还通过盐桥、ππ 相互作用等与周围的蛋白质残基相结合。
3 讨论
根据美国疾病控制和预防中心的报告[CDC,2007]在感染金黄色葡萄球菌的人群中,约1%的感染是由MRSA引起的。耐甲氧西林金黄色葡萄球菌对目前很多广谱抗生素都表现出耐药性[11-12],对公众构成严重威胁。本课题组研究的天然产物都是具有抗菌或者抑菌活性的,虽然其没有进行过抗MRAR 研究,但是只要PBP2a 蛋白的变构位点被一个药物分子占据,蛋白活性位点就会被打开,这些药物分子就可能成为抗MRAR的抗生素。
本研究通过分子对接研究,发现有Dihydrocalodenin B、Calodenin B、Piperaduncin C 等 24个化合物在变构位点和活性位点都有较低的结合能,预示着这24 个化合物可以先有一个分子与变构位点结合,触发PBP2a 构象变化,使蛋白的活性部位可以与另一个分子结合,从而达到抗MRAR 的作用。实际上,PBP2a表现出耐药性,是因为在PBP2a的变构位点(146位和150位)上发生了两个氨基酸的变化,这两个氨基酸的变化对头孢类药物产生了抗药性,导致了头孢类药物耐药MRSA 菌株的增加。本研究发现24 个化合物中有16 个与146 位通过非共价键相结合,例如,表1 和表2 中结合能都比较低的Dihydrocalodenin B、Calodenin B、Piperaduncin C 三个化合物在 3ZG0 变构位点上都与146位蛋白通过范德华力或者氢键结合,使其进入活性位点。本课题的研究结果将有助于设计出对耐药MRSA 菌株具有潜在治疗作用的有效化合物。
不良的ADMET 特性对有效药物开发的失败率有显著影响。由于ADMET 的研究在有效抗生素的开发中起着至关重要的作用,因此本研究也分析了所选化合物的理化性质。 结果发现Abyssinone V、Glycycoumarin 等9 个化合物表现出良好的ADMET 特性。值得注意的是,Isogancaonin C 水溶性较好,并且血脑屏障渗透能力也比较好。
有趣的是,在9 个化合物中,Gancaonin C、Glycycoumarin、Lupiwighteone 来自同一个中药甘草。现代研究表明,甘草的醇提取物在体外对金黄色葡萄球菌、结核杆菌、大肠杆菌、阿米巴原虫及滴虫均有抑制作用[13]。同样地,Kuwanon A 来自中药桑白皮,研究表明,桑白皮的煎剂对金黄色葡萄球菌、伤寒杆菌、福氏痢疾杆菌有抑制作用[14]。Quercetin-3-methyl ether来自中药青蒿,青蒿提取物对表皮葡萄球菌、卡他球菌、炭疽杆菌、白喉杆菌有较强的抑菌作用,对金黄色葡萄球菌、绿脓杆菌、痢疾杆菌、结核杆菌等也有一定的抑制作用[15]。Gancaonin C[16]、Glycycoumarin[17]、Lupiwighteone[18]、 Kuwanon A[19]、 Quercetin-3-methyl ether[20]均有较强的抗菌作用,值得进一步研究。

?

?

图1 表1和表2中化学成分的结构

表3 具有良好ADMET特性的9个化合物
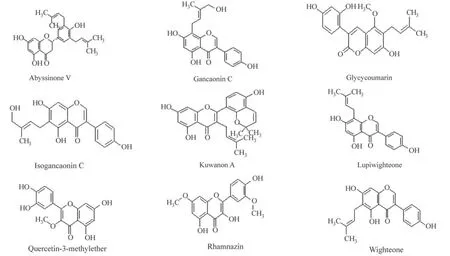
图2 表3中化学成分的结构
