利用CRISPR/Cas9系统构建人源结肠癌SAMHD1基因敲除细胞株及其功能的初步研究
2020-04-18张洲操孙润武晋英马孟涛宋晓宇
张洲,操孙润,武晋英,马孟涛,宋晓宇
(中国医科大学转化医学研究院,沈阳 110122)
结肠癌是世界上最常见的恶性肿瘤之一,其发病率已居全球第3位,死亡率也位于恶性肿瘤前列[1]。由于结肠癌细胞具有快速增殖能力和转移能力,结肠癌的发病率和致死率呈逐年上升趋势。近年研究表明,免疫基因在结肠癌发生发展中起重要作用,探讨免疫相关基因和蛋白在结肠癌细胞中的功能和作用,对结肠癌的防治具有重要意义。包含SAM和HD结构域的1型蛋白(sterile alpha motif and histidine-aspartic acid domain containing protein 1,SAMHD1)是一种脱氧核苷酸三磷酸水解酶,在维持细胞脱氧核糖核苷酸稳态平衡中发挥重要作用[2-3]。SAMHD1蛋白首次发现于树突状细胞,细胞SAMHD1基因突变会导致机体产生严重的免疫性疾病,如Aicardi-Goutières综合征、系统性狼疮、脑萎缩、癌症等[4]。常间回文重复序列丛集(clustered regularly interspaced palindromic repeat,CRISPR)/常间回文重复序列丛集关联蛋白9(CRISPR-associated protein,Cas9)系统是细菌和古细菌形成的一种免疫防御系统,利用单向导RNA(single-guide RNA,sgRNA)介导的Cas9 蛋白对基因组DNA进行特异性切割,从而达到更好的基因编辑作用,可帮助研究人员了解疾病分子机制[5-7]。为了探究SAMHD1蛋白在结肠癌细胞中的生物学功能和分子机制,本研究通过CRISPR/Cas9系统敲除SAMHD1基因,构建了SAMHD1基因敲除的HCT116人源结肠癌细胞株,进一步检测SAMHD1对结肠癌细胞增殖能力的影响,为深入研究SAMHD1在结肠癌细胞中作用的分子机制提供可选模型和理论基础。
1 材料与方法
1.1 主要材料和试剂
pYSY-CMV-Cas9-EF1α-Puromycin质粒购自南京尧顺禹生物科技有限公司;人结肠癌细胞(HCT116)购自于美国ATCC公司;T4 连接酶、E.coliDH5α购自日本TaKaRa公司;Tubofect transfection试剂购自美国Thermo Scientific公 司;anti-SAMHD1、β-actin抗体购自美国CST公司;琼脂糖凝胶DNA回收试剂盒、质粒小提试剂盒、无内毒素质粒中提试剂盒、细胞基因组DNA提取试剂盒、嘌呤霉素、IMDM培养基、限制性内切酶BbsⅠ均购自美国Sigma公司。
1.2 方法
1.2.1 人源SAMHD1基因结构分析及靶点设计:在美国国家生物技术信息中心数据库里找到人源SAMHD1的mRNA,根据实验目的选定目标结构域cd00077,找到该结构域对应的氨基酸序列中第一个M及核苷酸序列中对应的起始密码子,确定该起始密码子所在的外显子,输入预测sgRNA网站(http://crispr.mit.edu)进行预测。选取最高分引物:上游引物,5’-CACCGTATTCCACTTGCTCGCCCGG-3’;下游引物,5’-AAACCCGGGCGAGCAAGTGGAATAC-3’。在核酸序列nucleotide中找到sgRNA序列位置,并找到合适的鉴定引物位置,再使用DNACLUB进行检测,观察GC比例等。设计鉴定引物:上游引物,5’-GGCAACAAGAGCGAAACTCCGTCT-3’;下游引物,5’-CAGTGTCCAGGGTGCTTTCATTCACA-3’。
1.2.2 pYSY-CMV-Cas9-EF1α-Puromycin-sgRNASAMHD1重组质粒构建及克隆:使用限制性内切酶BbsⅠ酶切质粒使pYSY-CMV-Cas9-EF1α-Puromycin线性化,随后切胶回收。将合成的单链sgRNA常规退火后形成双链,用T4连接酶将之与纯化后的线性质粒连接。然后将连接产物转化到E.coliDH5α感受态中,涂于 AMP抗性的LB平板上。过夜培养后,挑取数个单菌落,送样测序,比对测序结果,选取含有正确的重组质粒的菌株扩大培养,进行中提取无内毒素质粒。
1.2.3 质粒转染及细胞培养:用含10%胎牛血清及青霉素(100 U/mL)和链霉素(100 U/mL)的IMDM 培养基,将HCT116细胞置于37 ℃、5%CO2孵箱进行培养,待细胞状态良好时将细胞接种于6孔板,约2.5×105/孔,设置Cas9-vector组和Cas9-SAMHD1组,各3个复孔。将9 μg Cas9-vector质粒和9 μg Cas9-SAMHD1重组质粒分别加入2个1.5 mL EP管,分别加入20 μL转染试剂Tubofect transfection和900 μL无血清培养基,混匀后分别取300 μL混合物,加入6孔板细胞中进行培养。转染48 h后,用嘌呤霉素1.5 μg/mL筛选,3 d后将筛选的混合细胞的1/2冻存,剩下的细胞继续培养,待状态良好,进行梯度稀释后接种于96孔板中,培养10 d后挑选单克隆细胞继续扩大培养。
1.2.4 TA克隆测序:取培养10 d后96孔板中的单克隆细胞置于48孔板、24孔板、6孔板,最后铺于10 cm培养皿中,取1/4提取基因组,然后进行PCR,采用TA克隆试剂盒对PCR引物进行TA克隆后送测序。
1.2.5 蛋白质印迹法检测SAMHD1蛋白水平:将选定的单克隆细胞扩大培养后,使用细胞裂解液(1%NP40)裂解细胞,提取蛋白并定量。利用SDSPAGE凝胶电泳分离蛋白后,采用恒压80 V电压转膜120 min,用5%牛血清白蛋白封闭液室温封闭1 h,加入一抗4 ℃过夜,次日TBST缓冲液洗膜3次,每次5 min,加入二抗室温孵育1 h,TBST缓冲液洗膜3次,每次15 min,采用ECL化学发光检测试剂盒检测蛋白水平。
1.2.6 检测DNA损伤指标γH2AX并采用CCK-8法和光学显微镜检测细胞增殖:分别将单克隆细胞传代至2个10 cm细胞培养皿,分别给予PBS(对照组)和0.5 mmol/L阿霉素(DNA损伤组),继续培养4 h后,进行蛋白质印迹实验,检测DNA损伤指标γH2AX。以2.5×103/孔的密度将细胞接种至96孔板,37 ℃、5%CO2培养箱过夜,待完全贴壁后,根据实验目的进行分组(Cas9-vector组和Cas9-SAMHD1组),每组3个复孔,培养24、48、72 h后,加入CCK-8试剂(5.0 g/L)10 μL/孔,置入细胞培养箱继续孵育1 h,酶标仪检测450 nm处各组吸光度,并比较各时间点细胞数量。将Cas9-vector组和Cas9-SAMHD1组的3个复孔450 nm处的吸光度值取平均值,用来表示细胞增殖能力的变化。
1.3 统计学分析
2 结果
2.1 利用CRISPR/Cas9系统打靶成功构建靶向敲除SAMHD1的CRISPR/Cas9载体体系
利用 BbsⅠ酶对pYSY-CMV-Cas9-EF1α-Puromycin进行线性化处理后,进行琼脂糖凝胶电泳,见图1A。将合成的sgRNA-SAMHD1引物配制成100 mmol/L的保存浓度,经过退火后,进行琼脂糖凝胶电泳,见图1B。待成功后,连接到已线性化的pYSY-CMV-Cas9-EF1α-Puromycin,并测序。测序结果表明,重组质粒构建成功,见图1C。
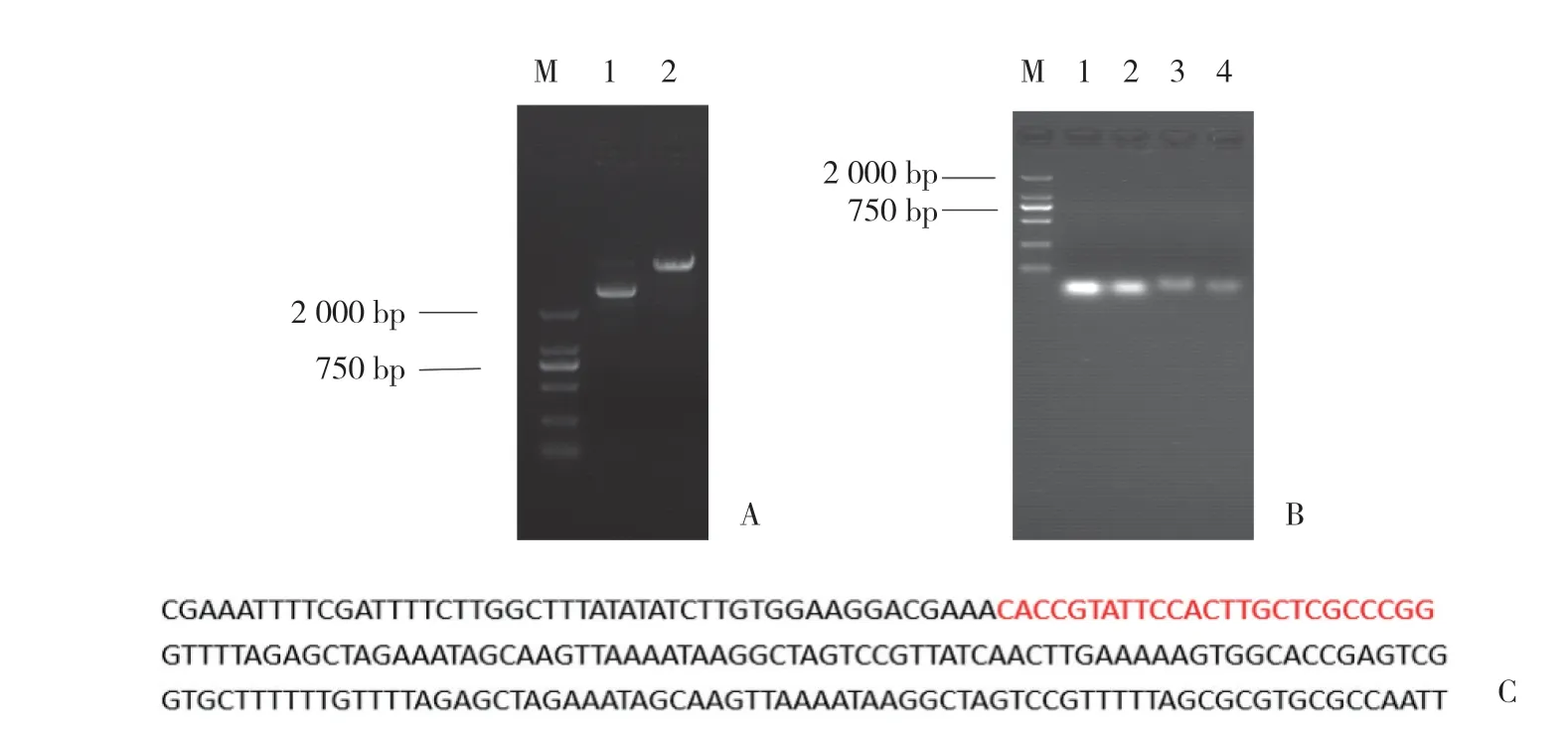
图1 利用CRISPR/Cas9 系统打靶构建基因敲除质粒图Fig.1 Images of gene knockout plasmids by CRISPR/Cas9 system targeting technology
2.2 利用CRISPR/Cas9系统成功构建敲除SAMHD1基因的HCT116细胞株
通过蛋白质印迹法检测敲除SAMHD1基因后Cas9-SAMHD1组和Cas9-vector组中HCT116细胞的SAMHD1蛋白表达水平,见图2。结果证明,敲除SAMHD1基因后Cas9-SAMHD1组SAMHD1蛋白表达水平在72×103位置条带亮度明显较低,SAMHD1蛋白的相对表达量为0.026±0.003;而Cas9-vector组SAMHD1蛋白的相对表达量为0.064±0.006,表明靶向敲除SAMHD1基因的HCT116 Cas9-SAMHD1细胞株构建成功。
2.3 验证敲除SAMHD1基因的稳定HCT116细胞DNA损伤修复能力较对照组细胞增强
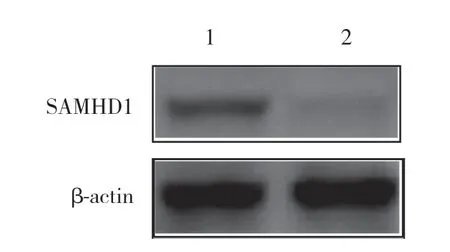
图2 Western blotting检测HCT116细胞中SAMHD1蛋白的表达Fig.2 Western blotting results of SAMHD1 protein expression in HCT116 cells
通过蛋白质印迹法检测Cas9-SAMHD1组和Cas9-vector组DNA损伤指标γH2AX的表达水平,见图3。结果显示,Cas9-SAMHD1组的γH2AX表达水平在15×103位置条带亮度明显较低,未给予阿霉素时和给予阿霉素4 h后γH2AX蛋白的相对表达量分别为0.140±0.013和0.201±0.007;而Cas9-vector组未给予阿霉素时和给予阿霉素4 h后γH2AX蛋白的相对表达量分别为0.119±0.006和0.465±0.016。结果表明,Cas9-SAMHD1组细胞抵抗DNA损伤的能力增强。
2.4 敲除SAMHD1基因后HCT116细胞的增殖能力较对照组显著增强

图3 Western blotting检测HCT116细胞中γH2AX蛋白表达Fig.3 Western blotting results of γH2AX protein expression in HCT116 cells
记录4个时间点(0、24、48、72 h)的细胞数量,放大倍数为10倍,见图4。光镜结果显示,Cas9-SAMHD1组细胞增殖能力较Cas9-vector组增强。计算细胞存活率,细胞存活率(%)=Cas9-SAMHD1组存活细胞数量/ Cas9-vector组存活细胞数量×100。Cas9-SAMHD1组与Cas9-vector组相比,细胞存活率的差异有统计学意义(均P< 0.01),见图5。CCK-8结果显示,Cas9-SAMHD1组细胞增殖能力较Cas9-vector组明显增强。

图4 HCT116细胞数量的变化 ×10Fig.4 Changes of HCT116 cells number ×10
3 讨论
本研究利用CRISPR/Cas9技术成功构建靶向敲除SAMHD1基因的HCT116细胞株,并证实该细胞株的增殖能力和抵抗DNA损伤的能力均较对照组显著增强。
从2012年起,CRISPR/Cas9系统被发展成为基因编辑工具,并被广泛用于多项科研和医疗领域。通过CRISPR/Cas9基因编辑技术可实现单个基因、位点的特异性突变,从而使特定基因完全丧失生物学功能[8-9]。通过利用sgRNA和Cas9蛋白,CRISPR/Cas9系统便可实现对目标DNA的识别和精确编辑。在很多方面该技术要明显优于其他基因编辑技术,尤其体现在其易于操作、效率高、周期短、工作量小、成本低等方面,并且可以在不同的位点同时引入多个突变,这是其他基因编辑技术不易做到的[10-11]。
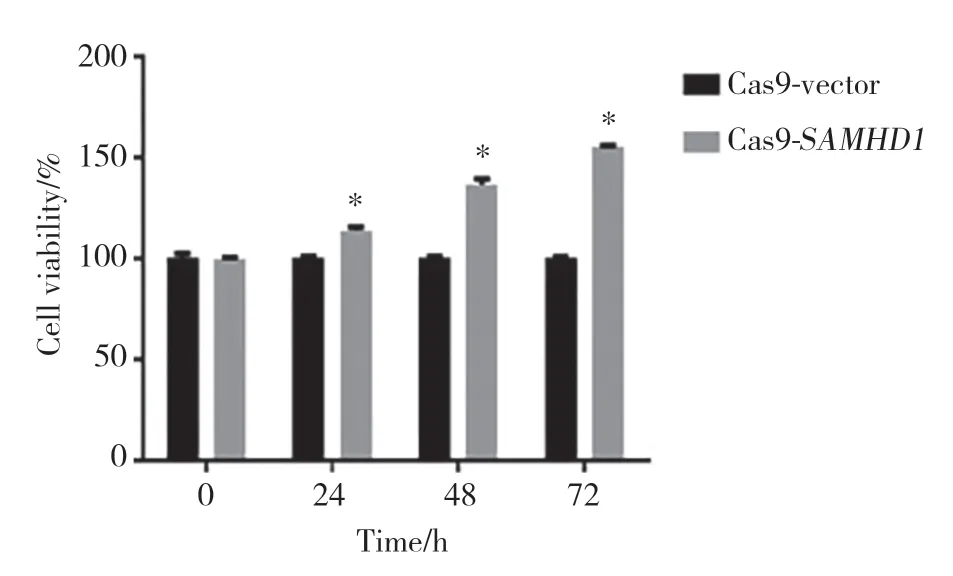
图5 HCT116细胞存活率的比较Fig.5 Comparison of survival rate of HCT116 cells
近年来,研究[12-15]证实SAMHD1是一种高效的dNTP水解酶,通过调控脱氧核糖核苷三磷酸的含量来维持基因组的稳定,并且可能通过降低细胞内的dNTP水平来抑制细胞增殖。研究[16]发现,SAMHD1在肺癌细胞中过表达后,可抑制核苷酸的形成和肺癌细胞的扩散,这提示SAMHD1可能与肿瘤细胞的增殖有关。与此同时,KODIGEPALLI等[17]发现,外源性SAMHD1表达通过促进细胞凋亡,在一定程度上抑制了皮肤T细胞淋巴瘤细胞的生长和增殖。这提示SAMHD1在肿瘤细胞生长中可能起抑制作用。本研究通过CRISPR/Cas9基因敲除技术成功敲除结肠癌细胞中SAMHD1,发现敲除SAMHD1后,结肠癌细胞的增殖活力显著增强,并且能够抵抗DNA损伤。
综上所述,SAMHD1基因参与调控肿瘤细胞增殖和DNA损伤修复反应。本研究结果有助于进一步了解SAMHD1在HCT116细胞以及其他肿瘤细胞中的生物学功能及分子机制。
