离子通道类镇痛药物作用机制及研究进展
2020-04-18于瑞河戚微岩刘晨高新梅徐寒梅
于瑞河,戚微岩,刘晨,高新梅,徐寒梅
(中国药科大学 江苏省合成多肽药物发现与评价研究工程中心,江苏 南京211198)
疼痛(pain)普遍存在于人类和多种动物中,是由直接或潜在组织损伤造成的一种令人不愉快的痛苦感觉,常伴有情绪的波动以及心血管和呼吸方面的变化[1]。而慢性疼痛(chronic pain)严重影响正常工作生活,加重患者经济负担,已成为一个主要的公共健康问题。传统镇痛药物包括吗啡(morphine)、芬太尼(fentanyl)等阿片类镇痛药(opioid analgesics)和水杨酸类、苯胺类等非甾体类抗炎药(nonsteroidal antiinflammatory drugs,NSAIDs)。虽然这些药物已被证实可以有效治疗疼痛,但因药物耐受性(tolerance)、成 瘾 性(addiction)、 消 化 道 出 血(digestive tract hemorrhage)等多重不良反应而受到限制[2]。因此,亟需开发出一种更为安全有效的新型镇痛药。近年来,研究者们在离子通道(ion channel)研究领域取得了突破性进展,研究表明抑制其相应的靶点具有明显的镇痛效果。本文聚焦于离子通道调控剂现状以及在缓解疼痛方面的应用,讨论目前离子通道类镇痛药物的作用机制及研究进展,希望能为科研工作者提供参考及思路。
1 离子通道概述
离子通道是一种含有水孔隙的封闭内在膜蛋白,可通过调控细胞内外离子流调节细胞膜上的电压电位[3]。离子流动产生电信号,以链式反应形式使相邻的电压敏感型通道开放,从而产生一种可以传导整个人神经细胞的自主传输电信号,传输速度约1m · ms-1。根据导致通道开放闭合的因素可将离子通道广义地分为电压门控离子通道(voltage-gated ion channel,VGIC) 和配体门控离子通道(ligand-gated ion channel,LGIC)。基于不同的离子通道位置和功能的不同,进一步将离子通道分为不同的亚型。研究表明,离子通道改变是炎症或神经病理损伤后外周敏化、中枢敏化及脱抑制作用的分子基础,也是疼痛发生的重要分子机制[4]。人类基因组中存在215 个离子通道编码基因,其中85 个与疼痛紧密相关,但仅有少数可用于疼痛治疗,已成功用于疼痛治疗的离子通道则更少。
近年来,借助基因突变手段已证明了离子通道在疼痛发生中的作用。研究表明在突变个体中,电压门控钠离子通道Nav1.7 功能片段的缺失可导致痛觉失敏[4],而功能片段的获得会导致痛觉敏化[5]。
2 离子通道类镇痛药物及镇痛机制概述
目前,大部分离子通道类镇痛药物是10 年前发现,甚至有的已经有50 年历史。比如卡马西平(carbamazepine,1)是20 世纪50 年代发现的第1 代抗惊厥药物(anticonvulsants),临床用于三叉神经痛、癫痫和躁狂症[6]。这一时期的药物往往先通过模拟临床症状建立体内模型或者离体组织模型,从“表型”上探究药物效果,然后进一步探究配体与靶点蛋白间明确的相互作用。
卡马西平通过阻塞钠离子通道抑制持续的节律性电位,阻断信号在三叉神经核突触上的信号转导进而缓解疼痛反应[7]。在高浓度下卡马西平也能够调节钙离子通道(calcium channels)和γ-氨基丁酸(γ-aminobutyric acid,GABA)受体,这一广泛的离子通道抑制作用决定了其具有抗心律失常、抗抑郁、神经阻滞、镇静等广泛的适应证[7]。在这类局部麻醉药物(local anesthetics)中,还有另一种更经典的药物——利多卡因(lidocaine,2),临床用于麻醉外周组织、缓解急性疼痛以及治疗慢性疼痛[8]。但是,由于这些局部麻醉药的剂量相对较高,在阻断电压门控钠离子通道的同时也会阻断钾离子通道(potassium channels)和钙离子通道[9]。正如其他拥有多重药理作用的药物一样,这些非选择性药物的安全性和副作用也限制了它们的长期使用[10]。
综上所述,虽然非选择性钠离子通道阻滞剂已经在急性疼痛的治疗中使用多年,但由于其除了可以抑制伤害性感受器中的钠离子通道以外,还对心脏和中枢神经系统(central nervous system,CNS)产生作用,使得其在临床上的应用受限[7]。近年来,随着研究人员深入研究以及膜片钳技术的快速发展,离子通道类镇痛药物取得了突破性进展。
离子通道类镇痛药物靶点主要分为VGIC 家族和LGIC 超家族2 大类。其中,VGIC 包括电压门控钙离子通道(voltage-gated calcium channel,VGCC,Cav)、电压门控钠离子通道(voltage-gated sodium channel,VGSC,Nav)和电压门控钾离子通道(voltage-gated potassium channel,VGPC,Kv)等;LGIC 包括瞬时电压感受器(transient receptor potential,TRP)、N-甲基-D-天冬氨酸(N-methyl-Daspartic acid,NMDA)受体、嘌呤能受体P2X、酸敏感 离 子 通 道(acid-sensing ion channel,ASIC)、 烟碱样乙酰胆碱受体(nicotinic acetylcholine receptor,nAChR)和A 型γ-氨基丁酸受体 (type Aγ-aminobutyricacid receptor,GABAAR)等。图1 为上述几类离子通道类镇痛药物作用机制简图。
3 电压门控离子通道类镇痛药物
3.1 电压门控钙离子通道类镇痛药物
VGCC 是去极化介导的钙离子进入神经元细胞的主要通路,可调控神经递质释放、分泌、兴奋-收缩偶联与细胞内基因表达和物质代谢[11]。VGCC 是一种膜蛋白复合体,由α1、β、α2δ 及γ 4 个亚基构成。其中,α1亚基有10 种不同的类型,是构成VGCC 水孔隙的主要亚单位[12]。根据VGCC 结构功能特点及对阻滞剂敏感程度的不同,将其分为L 型、N 型、P/Q 型、R 型和T 型5 种类型,其中L 型、N 型、P/Q 型、R 型为高压激活(high-voltage-activated,HVA)钙离子通道,T 型为低压激活(low-voltage-activated,LVA)钙离子通道。目前研究者已经发现了编码不同类型VGCC 的10 种编码基因[11],如表1 所示。

表1 不同类型的电压门控Ca2+通道所对应的编码基因Table 1 Encoding genes of different subtypes of voltage-gated calcium channels
现有钙离子通道镇痛药物的靶点以N 型VGCC 为主,其中加巴喷丁(gabapentin,3)早期用于癫痫的治疗[13],目前也用于神经病理性疼痛的治疗,2002 年辉瑞公司的加巴喷丁获美国FDA 批准用于带状疱疹后神经痛(postherpetic neuralgia,PHN)的治疗。加巴喷丁作为一种亲脂性GABA,最初被认为是通过活化谷氨酸脱羧酶增加GABA 水平治疗癫痫。后来发现加巴喷丁的正确作用机制是与VGCC 的亚基α2δ 相互作用,而这些亚基主要位于新皮质、海马、杏仁核和脊髓[14]。在后续的药物发现中,辉瑞公司通过改造加巴喷丁提高其药代动力学特性得到普瑞巴林(pregabalin,Lyrica,4),并于2004 年获得美国FDA 批准用于治疗糖尿病性外周神经痛(diabetic peripheral neuralgia,DPN)以及PHN,如今,这一药物已成为治疗糖尿病、神经病变相关慢性疼痛的金标准[7,15]。
齐考诺肽(ziconotide,ω-MVIIA,Prialt)是目前唯一上市的N 型VGCC 选择性镇痛药物,于2004 年获得美国FDA 和欧盟批准用于鞘内注射治疗其他药物不能耐受或治疗效果不佳的严重慢性疼痛[16-17]。齐考诺肽是从致幻芋螺(Conus magus)的毒腺中分离得到的一种ω-芋螺毒素,是含有25 个氨基酸残基的小分子多肽,其中有6 个半胱氨酸残基两两成对形成3 对二硫键。齐考诺肽作用于N 型VGCC 的α1β 亚基水孔隙内部,可明显阻断钙离子内流,抑制致痛递质释放[16]。实验表明,齐考诺肽对10%甲醛所致的Ⅱ相疼痛、完全弗氏佐剂诱发的慢性炎性疼痛和脊神经结扎所致的神经病理性疼痛均有明显的抑制作用[18-19],镇痛效果优于吗啡,但对急性疼痛镇痛效果较差。此外,齐考诺肽长期给药不会出现耐受性问题,鞘内低剂量给药可明显减少全身毒性。临床研究发现,齐考诺肽对于癌症与获得性免疫缺陷综合征引起的严重疼痛、非恶性慢性疼痛和术后疼痛具有良好的镇痛作用,且未发生镇痛耐受[20-22],同时,对顽固性重度慢性神经性疼痛也显示出一定的镇痛效果,并且与阿片类药物联用可减少阿片类药物使用剂量,改善了单一用药产生的耐受性问题[23]。但是,在高剂量下齐考诺肽会导致嗜睡、行为失控、视力模糊、低血压和记忆障碍等不良反应,这可能由于N 型VGCC 在脑中也有广泛分布,因此会导致中枢神经系统严重的不良反应[24],再者,鞘内给药手术也会增加患者感染的风险。
来考诺肽(leconotide,CVID,AM336)是在齐考诺肽基础上开发的一种选择性阻断N 型VGCC 的类似物,可静脉注射,不良反应相对齐考诺肽较低,主要用于治疗癌症引起的慢性疼痛[25]。
T 型VGCC 存在于心肌细胞、平滑肌细胞以及中枢神经系统的多种神经细胞中,可调控钙离子流调节细胞阈电位,进而导致细胞去极化及其他钙离子、钠离子、钾离子通道的激活,阻滞其可缓解神经性疼痛、炎症性疼痛和内脏疼痛[7]。Z944(结构未报道)是一种新型口服选择性T 型VGCC 阻滞剂[26],对于T 型VGCC的选择性是非T 型VGCC 的150 倍,在Ⅰ期临床试验研究中均表现出了良好的安全性和耐受性,目前正处于Ⅱ期临床试验研究。zonisamide(5)是NeuroMed 公司研发的T 型VGCC 阻滞剂,早期用于抗癫痫,后用于慢性神经痛。Moore 等[27]通过数据库调研进行疗效评估,发现缺乏足够的证据表明zonisamide 可缓解各种神经病理性疼痛。目前部分在研和已上市电压门控钙离子通道类镇痛药物见表2。
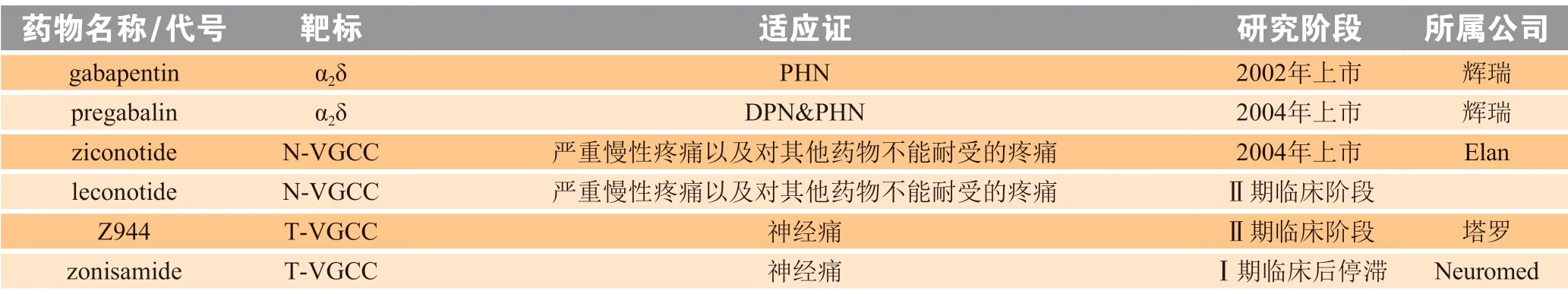
表2 目前部分在研和已上市电压门控钙离子通道类镇痛药物Table 2 Some of the analgesic drugs targeting voltage-gated calcium channel under clinical development or on the market
3.2 电压门控钠离子通道类镇痛药物
VGSC 是一类跨膜糖蛋白复合体,由一个α 亚基和数个β 亚基构成[28],广泛存在于可兴奋细胞(如神经元、骨骼肌细胞)的细胞膜上。在哺乳动物中,根据α 亚基的不同已鉴定出Nav1.1 ~ 1.9 9 种钠离子通道亚型[29]。其中,Nav1.1、Nav1.2、Nav1.3、Nav1.7 为河豚毒素敏感型(tetrodotoxin sensitive,TTX-S),Nav1.5、Nav1.8、Nav1.9 为河豚毒素不敏感型(tetrodotoxin resistant,TTX-R),与疼痛相关的亚型主要是Nav1.3、Nav1.7、Nav1.8、Nav1.9[33]。Nav1.3 在成年个体外围神经系统(peripheral nervous system,PNS)中表达量极少,仅少量存在于交感神经元[30],具体作用机制不明;Nav1.7主要存在于背根神经节(dorsal root ganglion,DRG)神经元、交感神经元[31]、施万细胞、神经内分泌细胞以及嗅上皮细胞中[32];Nav1.8 主要存在于三叉神经节神经元和DRG 神经元上[33];Nav1.9 主要分布于感受伤害的DRG 神经元[34]、三叉神经节和肠肌层神经元中[35];研究表明,Nav1.7、Nav1.8、Nav1.9 参与了炎症疼痛和神经疼痛过程[36-37],具体作用机制还在进一步研究。
利多卡因(lidocaine)是一种钠离子通道阻断剂,在局部神经病理性疼痛治疗方面作用稍差,神经受损后Nav1.7 和Nav1.8 的异常敏感被认为是疼痛产生的最重要因素,阻断Nav1.7 和Nav1.8 可减少痛觉神经纤维异位放电,提高外周异位放电阈值改善疼痛[38]。研究发现5%利多卡因药膏对外周神经源性疼痛和DPN 有良好的疗效[39-40]。
PF-05089771(6)为芳基磺酰胺类化合物,是一种正在进行临床试验并用于慢性疼痛的选择性钠离子通道阻滞剂,对Nav1.7 的选择性是Nav1.5 和Nav1.8 的1 000 多倍,并在临床Ⅰ期试验中表现出良好的耐受性。同时,从目前的研究现状来看,磺酰胺类(特别是芳基磺酸盐)几乎是开发Nav1.7抑制剂的主要化合物之一,这表明这类化合物可能比其他类具有更好的选择性优势[41]。此外,在临床开发中也存在其他类型化合物,比如Convergence 公司研发的吡咯烷基类化合物CNV-1014802(7),用于治疗三叉神经痛,目前正在进行Ⅲ期临床试验[42]。新型α-氨基酰胺类Na+通道阻滞剂 ralfinamide(NW-1029,8)在Ⅱ期临床试验中对多种疼痛模型及混合神经痛具有良好的治疗效果,但在Ⅲ期临床试验中对改善神经性下腰痛未表现出良好的治疗效果[43]。
除了小分子抑制剂外,一些对钠离子通道有选择性的天然毒素也用作疼痛的治疗,从狼蛛毒液中提取的protoxin Ⅱ[44]和从芋螺的毒液中提取的µ-Conotoxin-KⅢA[45]分别对 Nav1.7 和Nav1.4 有一定的特异性,用于治疗慢性神经痛等[46]。从少棘蜈蚣毒液中分离纯化出的小分子多肽μ-SLPTX-Ssm6a,作为一种Nav1.7 阻滞剂在甲醛致痛模型、醋酸扭体模型等多种经典镇痛模型和电生理实验中均表现出良好的镇痛效果[47]。另外,从河豚毒液中提取的河豚毒素(tetrodotoxin,TTX)可用于治疗与癌症相关的严重性疼痛[48]。
3.3 电压门控钾离子通道类镇痛药
根据钾离子通道作用机制的不同可将钾离子通道分为Kv、Ca2+激活型钾离子通道家族(KCa)、内向整流型钾离子通道家族(Kir)、双P 区(tandem pore domain)型家族4 种类型。其中,Kv7.x 属于延迟整流型钾离子通道,含6 个跨膜结构域和1 个空区,主要分布于交感神经节、DRG 神经元及部分中枢神经系统以及与癫痫发作有关的脑区[49]。研究表明,瑞替加宾(retigabin,9)作为一种Kv7.2 ~ 7.5 开放剂,可有效治疗神经结扎模型所产生的神经病理性疼痛、甲醛所导致的炎症疼痛,减缓辣椒素引起的内脏疼痛[50]。然而,Ⅱ期概念验证试验(proof-of-concept,PoC)中发现瑞替加宾在PHN 治疗中与安慰剂无明显差异。令人欣慰的是瑞替加宾的结构类似物氟吡汀(flupirtine,10)在临床试验研究中表现出较好镇痛活性[51]。
4 配体门控离子通道类镇痛药物
TRP 是一类存在于细胞膜上的非选择型阳离子通道家族,包含28 个成员,根据其氨基酸同源序列的不同可分为瞬时感受器电位香草酸受体(transient receptor potential vanilloid,TRPV,即辣椒素受体)、瞬时受体电位阳离子通道蛋白(transient receptor potential cation channel,TRPC)、瞬时受体电位通道(transient receptor potential melastatin,TRPM)、 瞬 时 受 体 电位 多 囊 蛋 白(transient receptor potential polycystic,TRPP)、瞬时受体电势阳离子通道粘脂蛋白(transient receptor potential cation channel,mucolipin subfamily,TRPML)、瞬时受体电位通道锚蛋白(transient receptor potential ankyrin,TRPA)6 个不同的亚族,其中与疼痛相关的亚基为TRPV1、TRPV3、TRPV4、TRPM3、
TRPM8、TRPA1[52]。
TRPV1 可温度被大于42℃的毒热、刺激性分子(如辣椒素)、内源性脂类分子等激活,一些TRPV1 阻滞剂已用于临床疼痛的治疗,比如AMG517(11)[53],SB-705498(12)[54]。然而,许多临床研究报告显示TRPV1 阻滞剂对核心体温有不良影响,这阻碍了针对该通道药物的进一步开发。
TRPV3 可被天然产物樟脑、2-氨基乙酯二苯基硼酸(2-aminoethoxydiphenyl borate,2-APB)和大于32℃的温度激活,例如从水蛭中分离的喹唑啉(quinazolin-4-one)[55]和Glenmark 公司的GRC15300(结构未报道),这2 种TRPV3 阻滞剂均具有较好镇痛活性。其中,Glenmark 公司已于2011 年完成了GRC15300 的Ⅰ期临床研究,并于2012 年转让给赛诺菲-安万特公司(Sanofi-Aventis)开展该药物在治疗神经病理性疼痛方面的Ⅱ期临床试验,但后者已于2014 年宣布该药物在Ⅱ期PoC 研究中失败[7]。
TRPV4 可被一些小分子化合物(大麻素等)和27 ~ 34℃的温度刺激而活化,近年来,已经发现了一些TRPV4 选择性阻滞剂,如RN-1734(Renovis,13)、HC-067047(Hydra,14)[56]。目前,只有一种未公布结构的GSK2798745 进入Ⅰ期临床[7],或将用于肺水肿引起的炎症疼痛。
TRPM8在体外可被10 ~ 23℃的温度或冷却剂(icilin或薄荷醇)活化。辉瑞公司已经公布了一种TRPM8 的拮抗剂PF-05105679(15),该拮抗剂被证明在人类冷痛实验模型(冷加压试验)中有良好的镇痛效果,并对人体核心温度无不良影响[57]。
TRPA1 可被外源性亲电体桂皮醛(cinnamaldehyde)、异硫氰酸烯丙酯(allyl isothiocyanate)和内源性配体羟壬烯醛(4-hydroxynonenal,4-HNE)等不同的配体激活,重组TRPA1 可被低于17℃的温度活化[58],TRPA1通道基因突变与人类家族性发作性疼痛有直接的联系。目前,发展最快的是Glenmark 公司的TRPA1 拮抗剂GRC17536[59](结构未报道),Ⅱa 期临床试验表明对糖尿病性神经病变具有明显的治疗作用。Hydra/Cubist公司研发的TRPA1 拮抗剂CB-625 也已完成了口腔疼痛和炎症疼痛的Ⅰ期临床试验,但具体数据还未公布[58]。
N-甲基-D-天冬氨酸(N-methyl-D-aspartic acid,NMDA)受体是一种离子型谷氨酸受体,广泛表达于DRG 神经元,其可允许钠离子、钾离子、钙离子通过,在病理性疼痛和痛觉过敏中起到重要作用[60]。NMDAR 具有多个结合位点,是调节慢性疼痛及与之相关神经毒性作用的关键靶点[61]。Traxoprodil(16)是一种NMDAR 亚型GluN2B 受体拮抗剂,已用于改善中枢神经失调,进入卒中和神经性疼痛治疗的临床研究阶段[62]。氯胺酮(ketamine,17)是美国FDA 批准的NMDAR 拮抗剂,可与NMDAR 相互作用阻滞兴奋性神经递质传递发挥麻醉作用,拮抗兴奋性氨基酸与NMDAR 结合从而抑制兴奋性突触后电位的产生和伤害性刺激的传入而发挥镇痛作用[63]。N2O(又称笑气)作为一种镇痛、抗焦虑的吸入性麻醉药物,临床应用已经有150 多年的历史,上世纪90 年代,N2O 首次被发现是作为一种NMDAR 离子通道拮抗剂[63]。
嘌呤受体P2X 是存在于神经元、免疫细胞、肿瘤细胞上的ATP 配体门控阳离子通道。近年来,P2X3、P2X2/3、P2X4 和P2X7 受体已作为一个潜在靶点用于治疗慢性疼痛和关节炎[64]。CE-224535(18)和AZD9056(19)是2 种P2X7 阻滞剂,已经分别由辉瑞和阿斯利康公司推向治疗类风湿关节炎的临床研究,遗憾的是均没有明显效果,目前关于针对疼痛治疗方面正进行深入研究[7]。
ASIC 属于上皮细胞钠离子通道/退化蛋白(epithelial sodium channel/degenerin,ENaC/DEG)超家族中的一员,其功能片段由ASIC1、ASIC2、ASIC33 个编码基因编码,分为ASIC1a、ASIC1b、ASIC2a、ASIC2b、ASIC3 5 个亚基,这些亚基以不同的组合方式结合形成pH 敏感性、离子选择性等不同的功能性异聚体通道。不同的亚型在PNS 和CNS 中的分布具有一定的特异性,ASIC 1b 和ASIC 3 主要分布于感觉神经元,ASIC 1a、ASIC 1b 和ASIC 2b 广泛分布于PNS 和CNS[65]。ASIC可调节神经病理疼痛和炎症性疼痛传导,酸性物质或低pH 环境都会激活ASIC,诱导炎症介质的反应,进而引发疼痛[66]。从海葵中分离得到的毒素APETx2 作为一种ASIC 抑制剂,能够与ASIC3 特异性结合从而发挥镇痛活性;另外从黑曼巴蛇毒中分离得到的多肽mambalgin-1也可以抑制PNS 和CNS 的ASIC,进而发挥镇痛作用[66],目前尚未见该类药物的进一步研究报道。
nAChR 属于Cys-loop 超家族,是由5 个亚基组成的跨膜五聚体的配基门控阳离子通道,具有多种亚型。在人类中含有α1~ α7、α9、α10、β1~ β4、δ、ε、γ 不同的亚基,分别由16 种基因编码,其中α1、β1、δ、ε、γ 亚基组成神经肌肉接头处的nAChR 亚型,其余的α 和β亚基以多种组合形式组成众多不同的受体亚型表达于神经元上[67]。α-芋螺毒素(alpha-conotoxin,α-CTx)是最早发现的一类芋螺毒素,一般由12 ~ 30 个氨基酸残基组成,富含二硫键,能特异性作用于神经型或肌肉型nAChR 发挥镇痛活性[68]。近年来,研究者陆续发现了可作用于nAChR 的新超家族或新家族芋螺毒素(α*-CTx),α*-CTx 能特异地作用于多种乙酰胆碱受体亚型,可作为新药开发的先导物深入研究[67-68]。
GABAR 是CNS 最主要的抑制性神经传导复合体,其中GABAAR 在中央杏仁核(CeA)中有很高的表达,介导前脑大多数快速突触抑制[69]。抑制性神经递质GABA 的作用与兴奋性递质谷氨酸相反,GABAAR 激动和 GABA 水平升高有利于镇痛[70],在已有麻醉药中苯巴比妥(phenobarbital,20)可提高神经系统GABA 水平[71]。GABAR 的另一种亚型GABABR 与长时程反射抑制作用密切相关,研究发现Vc1.1 可通过激动GABABR 来发挥镇痛活性[72]。
5 结语
疼痛是全球面临的重大公共健康问题,给患者的生活、工作、经济带来沉重的负担。早期的镇痛药物研发多以阿片受体为研究靶点,虽然针对这一靶点的镇痛药物具有很强的镇痛作用,但其存在毒性大和易成瘾的问题,不能满足临床对镇痛药物安全性、有效性和耐受性的需求。规避阿片类镇痛药物的毒性和成瘾性,积极探索治疗疼痛的新靶点、探明镇痛药物作用机制一直是研究人员追寻的目标。
离子通道类镇痛药物因具有良好选择性、耐受性和有效性以及不易成瘾等特性被广泛研究。其中,加巴喷丁、齐考诺肽等一些药物已经先后成功上市,还有一部分药物因在临床试验中有效性低而未能成功上市。造成这种现象的原因可能是:1)药物靶点分布在动物和人体间存在一定差异,有些疼痛的发生机制还不明确,药物发挥镇痛作用的机制有待完善;2)药物对靶点的选择特异性不高,易产生不良反应等。相信通过提高离子通道克隆与筛选能力,完善靶点选择性分子理论基础和镇痛机制,探索特异性离子通道与疾病间的内在机制联系,进而明确镇痛机制,寻找最佳作用靶点,不断优化,在未来会有更多的离子通道类药物走向临床,造福广大患者。
