富马酸丙酚替诺福韦合成工艺改进
2020-04-10赵明礼柴雨柱朱春霞
赵明礼,王 喆,舒 伟,柴雨柱,徐 丹,朱春霞
(南京正大天晴制药有限公司,江苏 南京 210046)
富马酸丙酚替诺福韦(1)是由美国吉利德科学公司研发的新型抗乙肝病毒药物,于2016年11月获美国FDA批准上市,是近10年内被批准用于治疗慢性乙肝的首种药物。2018年11月,1获得国家药品监督管理局批准上市。1的靶向性强,耐受性良好,服药剂量仅为上一代抗乙肝病毒药物替诺福韦二吡呋酯的十分之一即能达到同样的治疗效果[1-2]。
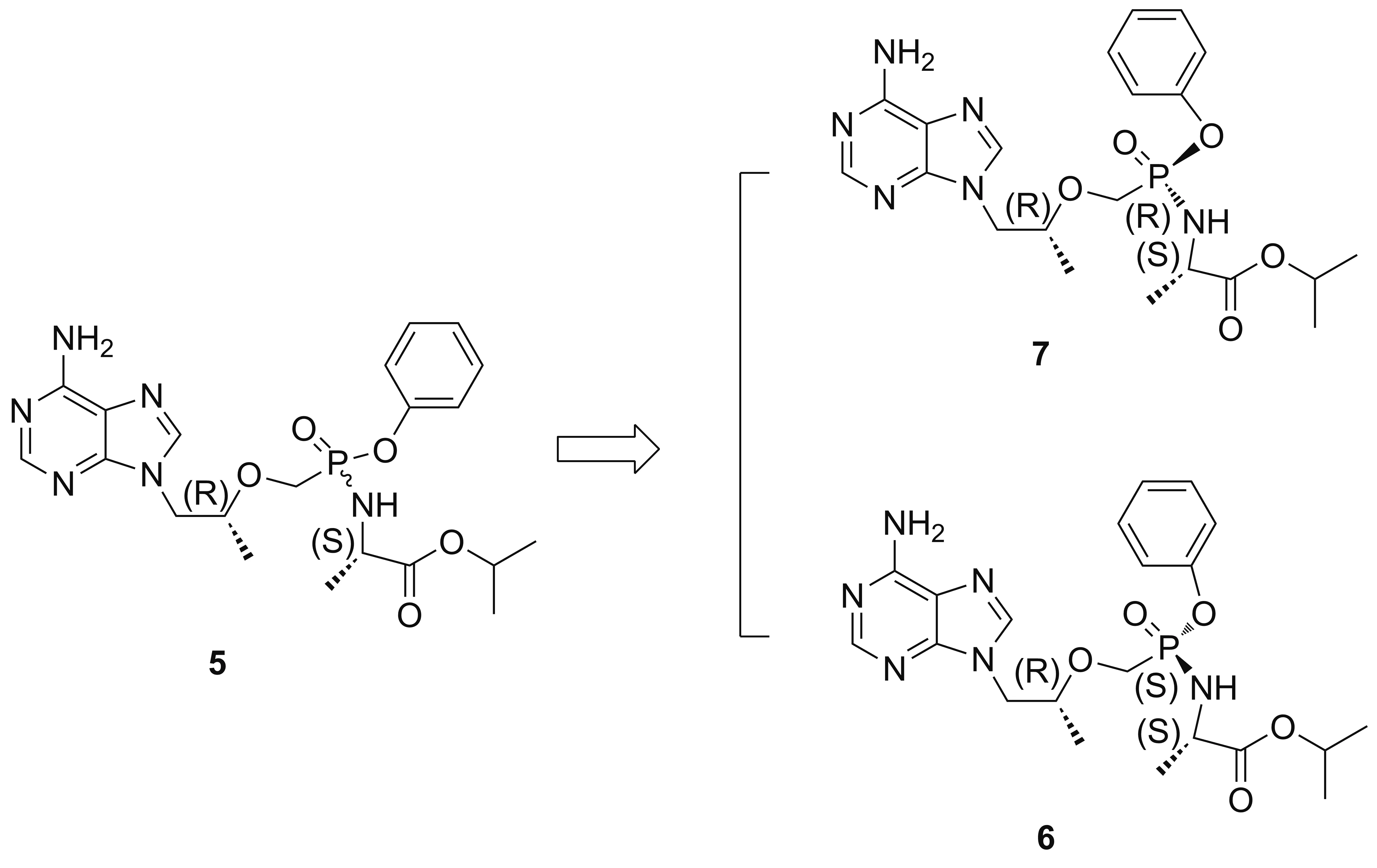
Chart 1
Scheme 1
近年来,有关1[3-18]及其类似物[19-22]的报道逐渐增多。1的合成均需要以替诺福韦(2)为起始原料:2首先与苯酚[5-6]或者亚磷酸三苯酯[7-18]反应制得(R)-9-(2-(苯基磷酰基甲氧基)丙基)腺嘌呤(3);3被氯代得化合物(R)-9-(2-(((苯基)(氯代)(磷酰基)甲氧基)丙基)腺嘌呤(4);4与L-丙氨酸异丙酯缩合得化合物9-((R)-2-(((S)-((1-(异丙氧基羰基)乙基)氨基)苯氧基磷酰基)甲氧基)丙基)腺嘌呤(5);5为一对非对映异构体,主要由9-((R)-2-(((S)-(((S)-1-(异丙氧基羰基)乙基)氨基)苯氧基磷酰基)甲氧基)丙基)腺嘌呤(6)和9-((R)-2-(((R)-(((S)-1-(异丙氧基羰基)乙基)氨基)苯氧基磷酰基)甲氧基)丙基)腺嘌呤(7)组成(Chart 1),经过手性柱分离或者拆分析晶可得到6,最后与富马酸成盐得到1。
现有合成1的方法主要存在以下问题:①化合物2容易吸水,形成稳定的水合物[16-17],影响化合物3的制备;②化合物4的合成有3种路线,分别以环丁砜、乙腈或者甲苯为溶剂,当使用环丁砜和乙腈为溶剂时[5-6],化合物5的纯度和选择性均较差,需要使用手性制备柱纯化分离,不适合工业生产;专利[7]报道使用甲苯为溶剂,反应48~96 h可得到90%以上非对映异构体纯度的化合物4,但重复时发现反应体系过于黏稠,搅拌困难,并且非对映异构体纯度仅约70%;③文献[7-13]中化合物5的合成均需要使用不稳定的L-丙氨酸异丙酯,工业化生产时需增加游离工序,操作复杂,成本较高。
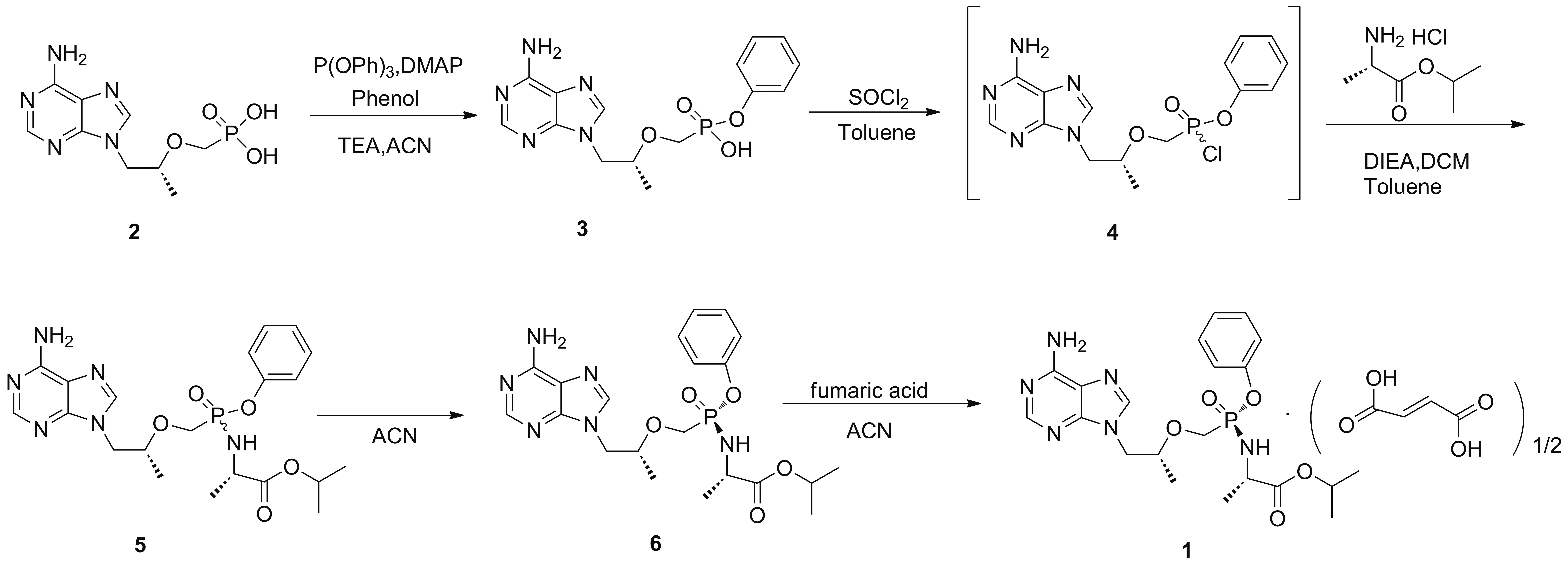
针对以上问题,本文对1的合成工艺进行了改进,以2为起始原料,与亚磷酸三苯酯反应得到3;3被氯化亚砜氯代得4;4与L-丙氨酸异丙酯盐酸盐缩合得5;5经析晶纯化得6;6与富马酸成盐得1(Scheme 1),其结构经1H NMR,13C NMR,MS(ESI),元素分析和XRD确证。
1 实验部分
1.1 仪器与试剂
Bruker AVANCE 3 HD 400/500 MHz型核磁共振仪(DMSO-d6或D2O为溶剂,TMS为内标);Waters Q-TOF MicroTM型质谱仪;Agilent 1200型高效液相色谱仪;PE 2400 Series II型元素分析仪;NETZSCH DSC 204型差热分析仪;Bruker D8 Advance 型X-射线衍射仪;Rudolph AUTOPOL VI型旋光仪。
化合物2,工业级(纯度≥99.0%),乐平市赛复乐医药化工有限公司;L-丙氨酸异丙酯盐酸盐,工业级(纯度≥99.0%),四川同晟生物医药有限公司;其余所用试剂均为工业级。
1.2 合成
(1) 化合物3的合成
氮气保护下,向50 L反应釜中加入化合物22.00 kg(6.96 mol)(提前干燥至水分≤0.5%)、三乙胺1.41 kg(13.93 mol)、4-二甲氨基吡啶850.73 g(6.96 mol)、亚磷酸三苯酯3.24 kg(10.45 mol)、苯酚655.35 g(6.96 mol)和乙腈16 L,搅拌下升温至80~85 ℃,保温反应48 h。浓缩,加入纯水6 L,用乙酸乙酯(3×6 L)萃取,合并水相,用浓盐酸调至pH 2~3,于0~10 ℃搅拌2 h。抽滤,滤饼于75~80 ℃鼓风干燥12 h得白色固体32.16 kg,收率85.3%,纯度99.20%(HPLC归一化法,下同);1H NMR(D2O)δ:1.26(d,J=9.7 Hz,3H),3.55(q,J=9.7 Hz,1H),3.81(q,J=9.7 Hz,1H),4.01~4.09(m,1H),4.25(q,J=9.7 Hz,1H),4.40(dd,J=9.7 Hz,1H),6.70~6.73(m,2H),7.05~7.09(m,1H),7.18~7.22(m,2H),8.21(s,1H),8.30(s,1H);13C NMR(D2O)δ:15.74,48.31,62.28,63.88,75.79,75.92,117.83,120.22,120.26,123.99,129.32,142.94,148.49,151.04,151.11,151.66,154.81;31P NMR(D2O)δ:14.88;MS(ESI)m/z:364.13{[M+H]+}。
(2) 化合物5的合成
向50 L反应釜中加入化合物32.10 kg(5.78 mol)和甲苯21 L,滴加氯化亚砜 1.51 kg(12.72 mol),滴毕,升温至65~75 ℃,保温反应48 h。浓缩,氮气保护下,残余物加入甲苯15 L,搅拌分散后降温至-20~-10 ℃得化合物4的甲苯溶液A。
氮气保护下,向100 L反应釜中加入L-丙氨酸异丙酯盐酸盐1.94 kg(11.56 mol),N,N-二异丙基乙胺2.99 kg(23.12 mol)和二氯甲烷18 L,降温至-20~-10 ℃,缓慢加入溶液A,控制内温为-20~-10 ℃,加毕,搅拌反应30 min。依次用5%磷酸二氢钠溶液(2×15 L),5%碳酸氢钾溶液(15 L)和纯水(15 L)洗涤,分液,有机相用无水硫酸钠2 kg干燥2 h。抽滤,滤液减压蒸除溶剂得红棕色油状液体52.70 kg,收率98.2%,化学纯度95.42%,非对映异构体纯度84.86%。未经纯化直接用于下一步反应。
(3) 化合物6的合成
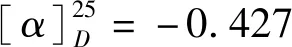
(4) 化合物1的合成
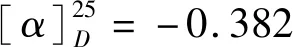
2 结果与讨论
2.1 化合物3合成条件的优化
表1为化合物3的合成条件优化结果。从表1可以看出,化合物2中的水分会影响该步反应,水分为5.3%时,收率78.0%,水分为2.7%时,收率为80.8%,水分为0.5%时,收率提高至85.3%,化合物3的纯度与水分没有明显关系。最终工艺在投料前对化合物2进行减压干燥,控制化合物2中水分≤0.5%。
2.2 化合物4合成条件的优化
表2为化合物4的合成条件优化结果。从表2可以看出,专利[7]报道的溶剂用量下反应搅拌困难,导致部分化合物3转化不完全,并且非对映异构体纯度仅70.53%,甲苯用量增大至10倍体积时,转化率及非对映异构体纯度均有增加,继续增加甲苯用量转化率及非对映异构体纯度没有明显变化;增加氯化亚砜用量后,转化率和非对映异构体纯度均得到进一步提高,考虑到工艺中需要浓缩氯化亚砜,规模化生产后会产生大量强酸性废液,所以未继续增大氯化亚砜用量;最后对比了反应时间对反应的影响,30 h化合物3已基本完全转化,非对映异构体纯度达到80%左右,延长至48 h,非对映异构体纯度达到85%左右,继续延长反应时间,非对映异构体纯度没有明显变化。最终工艺选择2.2 eq.氯化亚砜,10 倍体积甲苯,反应48 h。

表1 化合物3的合成条件优化Table 1 Optimization of the synthesis conditions of 3
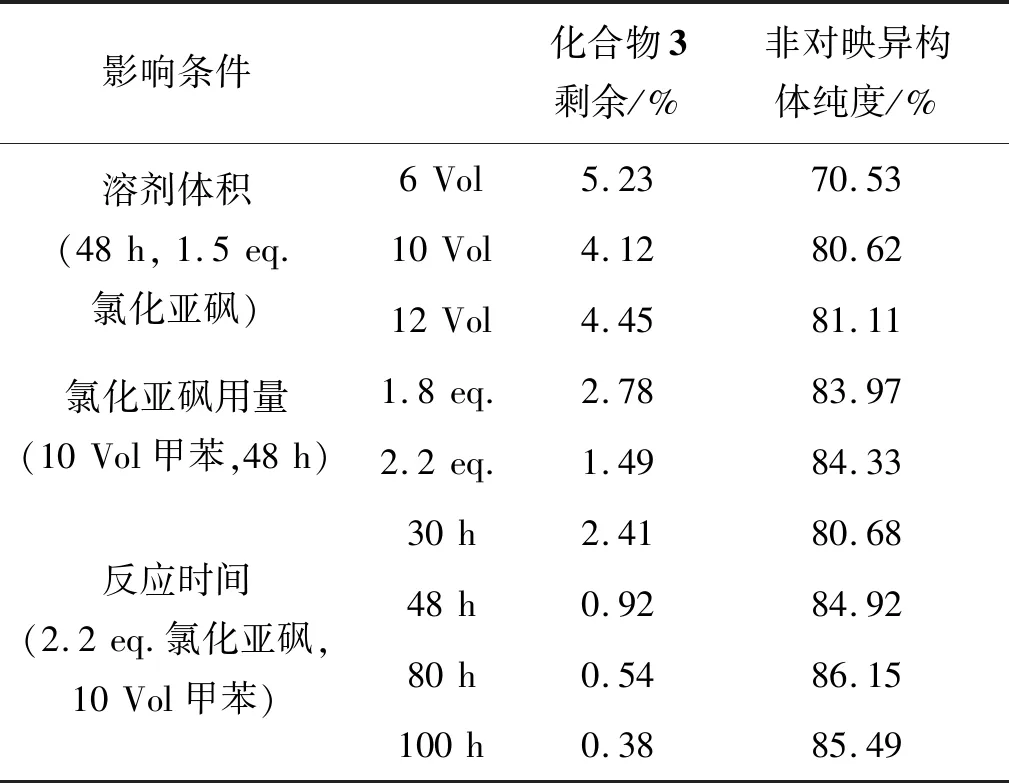
表2 化合物4的合成条件优化*Table 2 Optimization of the synthesis conditions of 4
*取反应液1 mL加入2 mL甲醇中摇晃淬灭,HPLC检测。
2.3 化合物5合成条件的优化
表3为化合物5的合成条件优化结果。从表3可以看出直接使用L-丙氨酸异丙酯盐酸盐与化合物4缩合,与使用L-丙氨酸异丙酯相比,收率和纯度没有明显变化。但使用盐酸盐,可避免游离操作,减少生产工序,降低生产成本,减少有机废液产生,降低污染。最终工艺选择使用L-丙氨酸异丙酯盐酸盐。
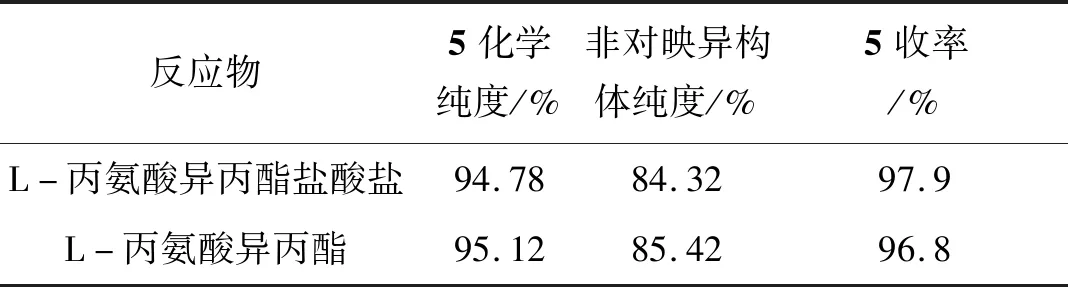
表3 化合物5的合成条件优化Table 3 Optimization of the synthesis conditions of 5
2.4 化合物6合成条件的优化
化合物6和化合物7在乙腈中溶解度差异较大,选用乙腈为溶剂对化合物5进行析晶纯化。考察析晶溶剂用量对析晶纯化效果的影响,第一次析晶效果如表4所示。由表4可见,使用2倍体积乙腈时,化合物5能够全部溶解,但是析晶时体系搅拌非常困难,不适合生产,未进一步研究;使用3倍和5倍体积乙腈时,均有一定的纯化效果,但是非对映异构体纯度均较低,这会导致在成盐工序无法得到非对映异构体纯度99.80%以上的产品,所以必须进行第二次析晶纯化。考虑使用3倍体积乙腈时收率较高,所以第一次析晶纯化选择3倍体积乙腈。

表4 化合物5第一次析晶效果Table 4 Result of the first crystallization of compound 5
表5为化合物5第二次析晶效果。由表5可见,第二次析晶纯化时,3~5倍体积乙腈纯化效果均较好,非对映异构体纯度均大于99.50%,且收率差异不大。3倍体积乙腈需较长时间才能完全溶解,在放大生产时可能会出现难以全部溶解现象,最后第二次析晶纯化选择4倍体积乙腈。
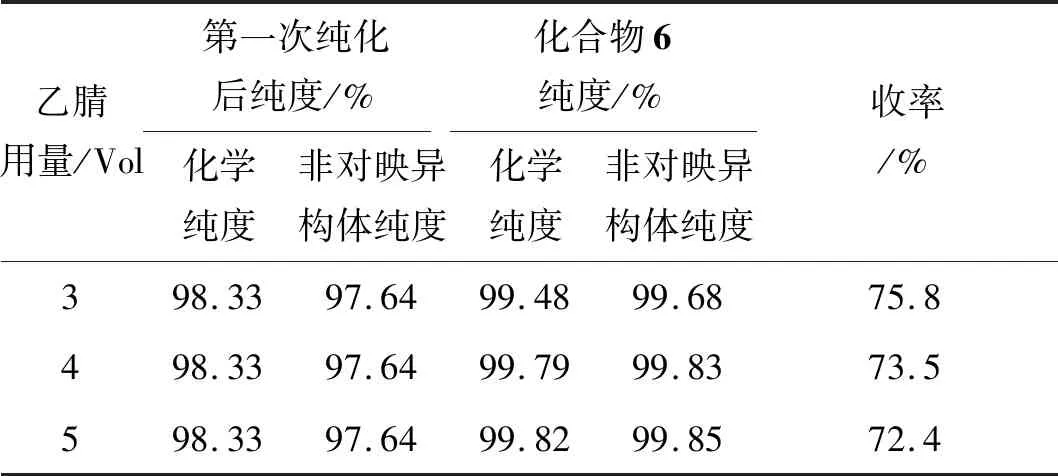
表5 化合物5第二次析晶效果Table 5 Result of the second crystallization of compound 5
2.5 化合物1合成条件的优化
表6为化合物1的合成条件优化结果。从表6可以看出,使用一次析晶得到的化合物6(化学纯度99.04%,非对映异构体纯度98.75%)与富马酸成盐,得到的化合物1的化学纯度为99.36%,非对映异构体纯度为99.47%,产品纯度较差,不满足药品生产需求;使用化学纯度为99.48%,非对映异构体纯度99.68%及以上纯度的化合物6与富马酸成盐,可以得到高纯度的化合物1。
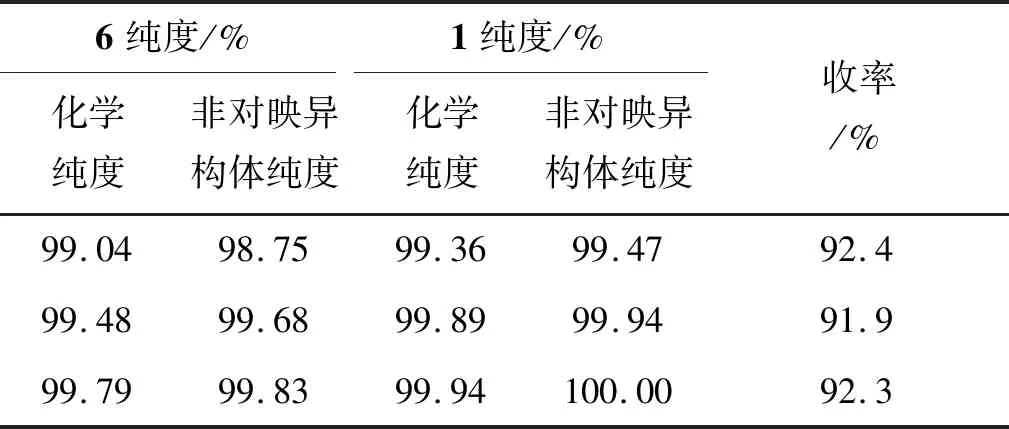
表6 化合物1的合成条件优化Table 6 Optimization of the synthesis conditions of 1
对富马酸丙酚替诺福韦合成工艺的关键参数进行了研究,并按照该工艺进行了公斤级规模放大。结果表明,该路线工艺稳定,操作简便,产品收率、化学纯度和非对映异构体纯度均较高,适合工业化生产。
