多方法制备草酸亚铁对其结构和形貌影响条件的探究
2020-04-08柯望贾艳梅余学峰吴慧敏王石泉孙争光
柯望,贾艳梅,余学峰,吴慧敏,王石泉,孙争光
(湖北大学化学化工学院,湖北 武汉 430062)
0 引言
草酸亚铁是一种化工原料,被广泛用于染料、涂料、陶瓷、玻璃器皿等的着色剂、新型感光材料的生产[1-2],也是合成纳米磁性材料、超级电容器的多孔材料及锂离子电池磷酸铁锂正极材料所需的主要原材料[3-4].随着我国科技的发展,智能手机、笔记本电脑、新能源汽车的需求日益增多,对作为合成锂离子电池磷酸铁锂正极材料铁源的草酸亚铁,需求量也在逐年增加.以草酸亚铁作为正极材料的铁源具有以下优点:1)草酸盐在合成过程中不易引入杂质相[5];2)草酸亚铁合成的磷酸铁锂正极材料结晶度较高且键合力大,有助于稳定合成产物的骨架结构[6];3)草酸亚铁在反应过程中会分解放出气体,可抑制颗粒的团聚和晶粒的长大[7].因此,与一般工业生产锂离子电池所用的正极材料层状结构的LiCoO2、LiNiO2材料、尖晶石结构的LiMn2O4材料等[8]相比,具有橄榄石结构的磷酸铁锂有原材料来源丰富、价格便宜、比容量高、热稳定性好、对环境友好等优点[9-12].
以草酸亚铁作为合成磷酸铁锂的原料时,草酸亚铁粒径的大小对合成的磷酸铁锂性能影响很大.粒径较为粗大的固体结晶在合成磷酸铁锂的固相反应过程中反应活性差,难与其它反应物混合均匀,使制得的磷酸铁锂正极材料颗粒粗大,导电性能差;当粒径较小(小于3.0 μm)时,草酸亚铁具有较高的比表面积,反应活性大大增加,合成出来的磷酸铁锂电化学性能较好[13-15].但是,综合目前国内工业生产草酸亚铁的情况来看,合成草酸亚铁技术尚且不够成熟,制备的草酸亚铁纯度较低、粒径大,在总的水平上和世界发达国家相比还有一定的距离[16].为了探究合成粒径较小的纯相草酸亚铁的生成条件,本文中以(NH4)2Fe(SO4)2·6H2O和 H2C2O4·2H2O为原料,采用多种合成方法:室温陈化法、水浴加热法、流变相法和溶剂热法合成草酸亚铁,并研究溶剂比例、温度、合成方法对草酸亚铁的结构和形貌的影响,探究合成尺寸较小、具有较好形貌的纯相草酸亚铁的实验条件.
1 实验部分
1.1 实验方法
1)室温陈化法.称取一定量的草酸和硫酸铁铵,加水溶解,用磁力搅拌器在室温下搅拌30 min,陈化3 h,待反应完全后,用蒸馏水和无水乙醇多次洗涤,进行固液分离,在60 ℃下干燥12 h,得到淡黄色的草酸亚铁产品.
2)水浴加热法.称取一定量的草酸和硫酸铁铵,分成4 份,分别加入水、体积比为3∶1的乙二醇(EG)和水、体积比为1∶1的EG和水、EG,用集热式恒温加热磁力搅拌器在90 ℃的条件下,搅拌3 h,待反应完全,用蒸馏水和无水乙醇多次洗涤,进行固液分离,在60 ℃下干燥12 h,得到淡黄色的草酸亚铁产品.
3)流变相法.称取一定量的草酸和硫酸铁铵,分成两份,分别置于研钵中,一份加入少量水,一份加入少量EG,在室温条件下,研磨30 min,然后将产物取出,用蒸馏水和无水乙醇多次洗涤,进行固液分离,在60 ℃的烘箱中干燥12 h,得到淡黄色的草酸亚铁产品.
4)溶剂热法.称取一定量的草酸和硫酸铁铵,将其分成3组,每组4份,向其中分别加入水、体积比为3∶1的EG和水、体积比为1∶1的EG和水、EG,用磁力搅拌器在室温下搅拌30 min,然后装入反应釜中,分别在120 ℃、150 ℃、180 ℃的烘箱中反应3 h,待反应完全,取出,用蒸馏水和无水乙醇多次洗涤,进行固液分离,在60 ℃的烘箱中干燥12 h,得到淡黄色的草酸亚铁产品.
综上,本研究合成草酸亚铁的化学反应方程式为:
(NH4)2Fe(SO4)2·6H2O+H2C2O4·2H2O=FeC2O4·2H2O↓+H2SO4+(NH4)2SO4+6H2O
1.2 样品的表征采用X线衍射(XRD)、红外(FT-IR)、扫描电镜(SEM)对产物进行结构和形貌表征.X线衍射采用日本(理学)D/max-rA X线衍射分析仪;红外采用Nicolet AVATAR 360型分析仪;SEM为日本电子(JOEL)公司JSM-650LV型号.
2 结果与讨论
2.1 XRD表征自然界中存在的草酸亚铁有2种不同的晶型:α-草酸亚铁和β-草酸亚铁.α-草酸亚铁属于单斜晶系,空间群为C2/c(No.15),晶胞参数为a=0.992 1 nm,b=0.555 6 nm,c=0.970 7 nm,β=104.5°;β-草酸亚铁属于正交晶系,空间群Cccm(No.66),晶胞参数为a=1.226 nm,b=0.557 nm,c=1.548 nm[17-19].下图1中为室温陈化法、水浴加热法、流变相法和溶剂热法合成的草酸亚铁晶体的XRD图.如图1(a)所示,可以观察到当陈化温度为室温时,所有的衍射峰均可标定为β-草酸亚铁的特征峰(PDF 22-0635),且未检索到其他杂质峰,经模拟计算其晶胞参数为a=1.222 nm,b=0.555 6 nm,c=1.544 nm,与β-草酸亚铁的晶胞参数相吻合,即室温陈化合成的样品为β型草酸亚铁;流变相法合成的样品也呈现出与室温陈化法相同的衍射峰,合成β型的草酸亚铁;当采用90 ℃水浴加热法合成样品的时候,发现晶型出现从β到α型转换,并且可以明显观察到α型草酸亚铁衍射峰(111)的出现.

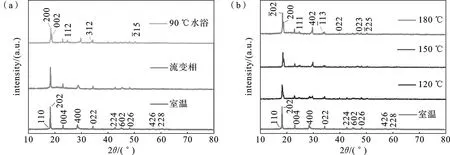
图1 (a)室温陈化法、水浴加热法(90 ℃)、流变相法合成草酸亚铁和(b)室温陈化法、溶剂热法(120、150、180 ℃)合成的草酸亚铁的XRD图
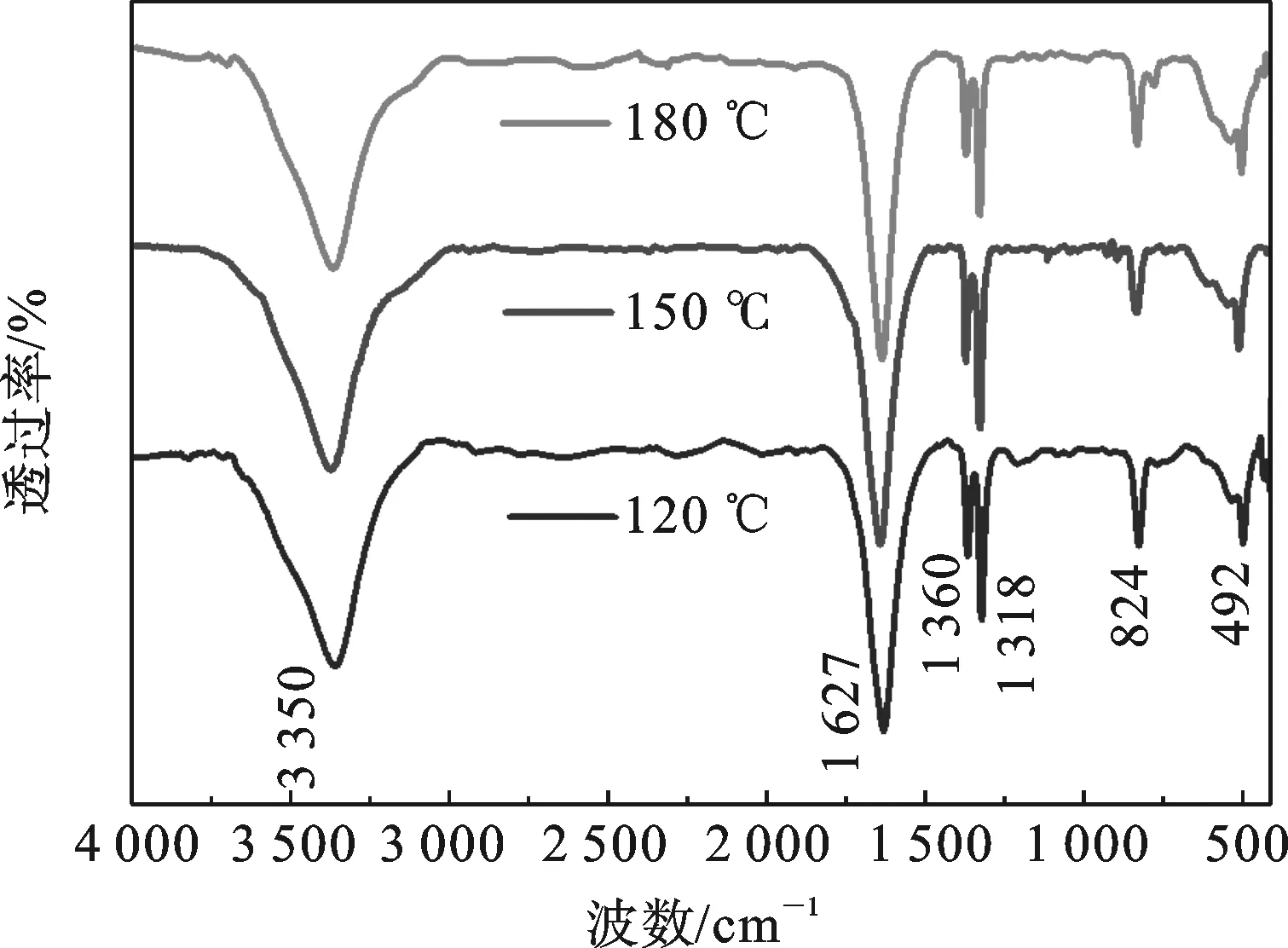
图2 不同温度下溶剂热法合成的草酸亚铁的红外光谱图
上述研究表明,β-草酸亚铁在热力学上处于不稳定的状态,随着温度的升高,β-草酸亚铁的晶体结构有向α-草酸亚铁变化的趋势.当温度越低,体系过饱和度就越大,粒子运动阻力大,扩散缓慢,进而晶体生长速度降低,易形成β-草酸亚铁;当温度升高的时候,粒子运动速度加快,晶体生长迅速,倾向于形成更加稳定的状态下的α-草酸亚铁.且衍射峰强度变化也体现了这一过程,从室温到180 ℃合成的样品的衍射峰强度发生了先变弱再变强的过程,这是由于其间晶型发生转换的缘故,最终在180 ℃时形成了衍射峰较强即结晶度较好的α-草酸亚铁.

2.2 SEM表征文中制备的所有草酸亚铁采用扫描电镜进行了微观结构的分析,以研究合成方法和溶剂对其粒径的影响.图3、图4为室温陈化法、水浴加热法、流变相法、溶剂热法合成的草酸亚铁的SEM图.图3为室温陈化法、水浴加热法、流变相法合成的草酸亚铁的SEM图.由图3(a)可见,室温陈化法合成的晶体呈现不规则块状,晶粒颗粒大小不均匀,晶体长度约4~7 μm,直径约2~4 μm.图3(b),(c),(d),(e)为水浴加热法合成的草酸亚铁的SEM图,从图中可以观察到,水浴加热法合成的样品整体分布不均匀,部分有棒状结构出现,有团聚现象,晶体长度和直径与溶剂有关.图3(f),(g)为流变相法合成的草酸亚铁的SEM图,图中显示流变相合成的样品晶体整体呈现较为均匀的颗粒状,团聚现象不明显.图4为溶剂热法合成的草酸亚铁的SEM图,从图中可以看到晶体整体形貌较好,具有明显的棒状结构,且粒径小,长度为2~4 μm,直径为200~500 nm.下文中将所有合成的草酸亚铁晶体的形貌粒径分布进行了统计制备成了表1,用以分析不同方法和溶剂合成对形貌粒径的影响.

图3 不同条件下合成的草酸亚铁的SEM图

图4 溶剂热法合成的草酸亚铁的SEM图

表1 不同方法下合成草酸亚铁晶体的粒径
通过表1可对晶体长度和直径的统计,结合SEM图发现,当溶剂采用体积比EG∶H2O=3∶1时,无论何种方法制备草酸亚铁,易于出现棒状结构,且形成了分布较为均匀的晶粒,这与溶剂的性质有关.根据热力学原理,粒子团聚是体系自由能降低的一种自发趋势,在结晶沉淀的过程中生成的粒子往往会通过静电引力或范德华力聚合在一起,以降低其巨大的表面能.而分散剂的添加可以降低产品颗粒表面的张力,减缓颗粒间的聚集和颗粒的沉降,抑制晶粒间的结合,使晶体生长缓慢,降低产品粒径,使晶粒不能相互吸附形成粗大的晶体,最后形成分布均匀的晶粒[21-22].EG作为一种功能强大的螯合剂,在反应中承担了分散剂的功能,在反应过程中,与过渡金属阳离子配合形成络合物,有效地减少了晶粒之间的团聚,降低了产品粒径,同时加入合适的量可有助于形成特定的棒状形貌.其参与反应的过程如下[23]:
M2++HOCH2CH2OH→(MHOCH2CH2OH)2+complexH2C2O4→2H++C2O42-(MHOCH2CH2OH)2++ C2O42-+2H2O→MC2O4·2H2O+HOCH2CH2OH
结合上述XRD分析结果,α型的草酸亚铁更具有热力学稳定性,故当采用溶剂热的方法,且在溶剂体积比为EG∶H2O=3∶1的情况下,制备出的晶体形貌最好,具有均匀的棒状结构,且粒径较小,分布较均匀.
3 结论
1)不同方法和反应温度下合成的草酸亚铁晶型不同,合成的β-草酸亚铁具有热力学不稳定性,逐渐向α-草酸亚铁转变.采用溶剂热法可在180 ℃时得到纯相的α-草酸亚铁.
2)溶剂比例对合成的草酸亚铁的形貌影响较大.当溶剂中EG比例增高,晶体粒径首先越来越小,但EG比例超过3∶1时,晶体形貌尺寸开始增大并有团聚现象;溶剂采用体积比EG∶H2O=3∶1时,无论何种方法制备草酸亚铁,易于出现棒状结构.
3)采用溶剂热法,在溶剂体积比为EG∶H2O=3∶1时,可以得到粒径小、具有棒状形貌的α-草酸亚铁晶体,为合成用于制备磷酸铁锂正极材料的草酸亚铁提供一条可行途径.
