Gitelman syndrome caused by a rare homozygous mutation in the SLC12A3 gene:A case report
2020-04-08RiZhenYuMaoShengChen
Ri-Zhen Yu,Mao-Sheng Chen
Ri-Zhen Yu,Mao-Sheng Chen,Department of Nephrology Division,Zhejiang Provincial People’s Hospital,People’s Hospital of Hangzhou Medical College,Hangzhou 310014,Zhejiang Province,China
Abstract BACKGROUND Gitelman syndrome(GS)is an unusual,autosomal recessive salt-losing tubulopathy characterized by hypokalemic metabolic alkalosis,hypomagnesemia and hypocalciuria.It is caused by mutations in the solute carrier family 12 member 3(SLC12A3)gene resulting in disordered function of the thiazidesensitive NaCl co-transporter.To date,many types of mutations in the SLC12A3 gene have been discovered that trigger different clinical manifestations.Therefore,gene sequencing should be considered before determining the course of treatment for GS patients.CASE SUMMARY A 55-year-old man was admitted to our department due to hand numbness and fatigue.Laboratory tests after admission showed hypokalemia,metabolic alkalosis and renal failure,all of which suggested a diagnosis of GS.Genome sequencing of DNA extracted from the patient’s peripheral blood showed a rare homozygous mutation in the SLC12A3 gene(NM_000339.2:chr16:56903671,Exon4,c.536T>A,p.Val179Asp).This study reports a rare homozygous mutation in SLC12A3 gene of a Chinese patient with GS.CONCLUSION Genetic studies may improve the diagnostic accuracy of Gitelman syndrome and improve genetic counseling for individuals and their families with these types of genetic disorders
Key Words:Gitelman syndrome;Hypokalemia;SLC12A3;Homozygous;Rare mutation;Case report
INTRODUCTION
Gitelman syndrome(GS),an autosomal recessive disorder caused by a defect in the gene coding for the thiazide-sensitive NaCl cotransporter at the distal tubule,is characterized by hyperreninemic hyperaldosteronism with normal or low blood pressure,hypokalemia,metabolic alkalosis,hypomagnesemia and hypocalciuria.It is usually diagnosed in late childhood or adulthood.Symptoms of hypokalemia include fatigue,leg cramps and constipation,but,most critically,slowing of the heart rhythm and even cardiac arrest.Affected individuals may experience episodes of fatigue,dizziness,fainting due to hypotension,muscle weakness,muscle aches,cramps and spasms or even tetany.Symptomatic episodes may also be accompanied by abdominal pain,vomiting,diarrhea or constipation,and fever.Seizures may also occur and in some people may be the initial reason they seek medical assistance.Facial paresthesia characterized by numbness or tingling is common.Tingling or numbness of the hands occurs less often.Affected individuals may or may not experience polydipsia,and polyuria including nocturia.When these symptoms do occur they are usually mild.Affected individuals often crave salt or high-salt foods.Some affected adults develop chondrocalcinosis which is thought to be related to hypomagnesemia.Affected joints may be swollen,tender,reddened,and warm to the touch[1].
TheSLC12A3gene,which is located on chromosome 16q13,encodes a transporter that mediates sodium and chloride re-absorption in the distal convoluted tubule of the kidneys[2].An inactivating mutation in the gene is a recognized diagnostic criterion for GS.GS is usually managed with oral potassium and magnesium supplements,potassium-sparing diuretics,spironolactone or occasionally nonsteroidal antiinflammatory drugs to control the symptoms in these patients[3].We present a 55-yearold man diagnosed with GS on the basis of his clinical features,laboratory tests and genetic analysis.We illustrate the management of his condition and review the relevant literature.
CASE PRESENTATION
Chief complaints
A 55-year-old man was admitted to the Emergency Department complaining of numbness in his hands and fatigue,without dizziness,vomiting or seizures.
History of present illness
The patient’s symptoms started 12 years ago with recurrent numbness in the upper limbs and fatigue.He was diagnosed with hypokalemia in a local hospital and took potassium supplements intermittently.His symptoms had worsened over the last 4 d.
History of past illness
He had been diagnosed with hepatitis B virus infection.
Physical examination
The patient’s temperature was 37.1 °C,pulse rate was 59 bpm,respiratory rate was 16 breaths/min,blood pressure was 111/88 mmHg,and oxygen saturation in room air was 97%.Physical examination findings were within the reference range.
Laboratory examinations
During the patient’s presentation in the Emergency Department,his serum chemistry revealed severe hypokalemia with a potassium level of 1.99 mmol/L(normal range:3.50-5.50 mmol/L).In addition to low potassium,his creatinine level was 138.6 μmol/L.
After symptomatic treatment,he was transferred to the Department of Nephrology for further clinical diagnosis and therapy.Laboratory analysis revealed a low potassium level(3.21 mmol/L),normal magnesium(0.93 mmol/L)and increased creatinine level(166.2 μmol/L).In addition,his liver enzyme levels were increased(aspartate aminotransferase 104 U/L and alanine aminotransferase 54 U/L)due to hepatitis B virus infection.Plasma renin activity and aldosterone level during rest periods were elevated at 18.331 ng/mL/h and 244.6 pg/mL,respectively.Hypoproteinemia and dyslipidemia were not detected.Urinalysis revealed protein 2+and occult blood 2+.Twenty-four-hour urine protein was 828.8 mg.Other serum and urine biochemistry findings are shown in Table 1.An electrocardiogram exhibited left ventricular high voltage and changes in the T wave.
Imaging examinations
A chest computed tomography scan revealed scattered calcifications.On ultrasonography,liver cirrhosis with multiple nodules and no pathology was seen in the spleen,pancreas and gallbladder.A urinary computed tomography scan showed multiple cysts and stones in the kidneys.
FINAL DIAGNOSIS
Written informed consent for genetic analysis was obtained from the patient.Total DNA was extracted from peripheral blood for whole genome sequencing(Joingenom Diagnostics,Hangzhou,China).A homozygous mutation T to A at position 536 in exon 4 ofSLC12A3gene(Figure 1)was identified in this patient.This rare mutation caused a Val to Asp substitution,resulting in a missense mutation affecting gene function.A similar novelSLC12A3pathogenic mutation was reported in a cohort of Chinese patients with GS previously[4].
TREATMENT
The patient was successfully treated with potassium chloride sustained-release tablets at a dose of 1 g three times/day.He was discharged from hospital 8 d after admission,and his potassium level was 3.5 mmol/L.He was maintained on potassium 1 g/d and spironolactone 20 mg twice/day.He was also given valsartan for proteinuria and entecavir for hepatitis B virus infection.
OUTCOME AND FOLLOW-UP
After discharge,the patient reported relief of numbness in his hands and fatigue and his serum potassium level remained in the normal range.
DISCUSSION
GS,also known as familial hypokalemia,is an autosomal recessive tubular disease[5].The typical clinical manifestations of GS patients are "five low and one high",which means hypokalemia,hypomagnesemia,hypochloremia,low urinary calcium,hypotension,and renin-angiotensin-aldosterone system(RAAS)activation[6].The common clinical manifestations of GS are mostly non-specific,often associated with electrolyte imbalance and increased RAAS activity[7].Even without long-term oral diuretics and other drugs,vomiting,diarrhea,hypercortisolism,and primary aldosteronism,GS can be diagnosed by clinical manifestations and laboratory tests alone.However,GS is easily misdiagnosed as classic Bartter Syndrome(BS)with analogous clinical features.Some researchers believe it is better to consider BS and GS as a spectrum of disease rather than distinct disorders.The defect in GS involves thedistal convoluted tubule while in BS the defect is in the thick ascending limb.Renal salt wasting is more severe and begins earlier in life in BS than in GS with manifestations rarely occurring in the neonatal period[8].
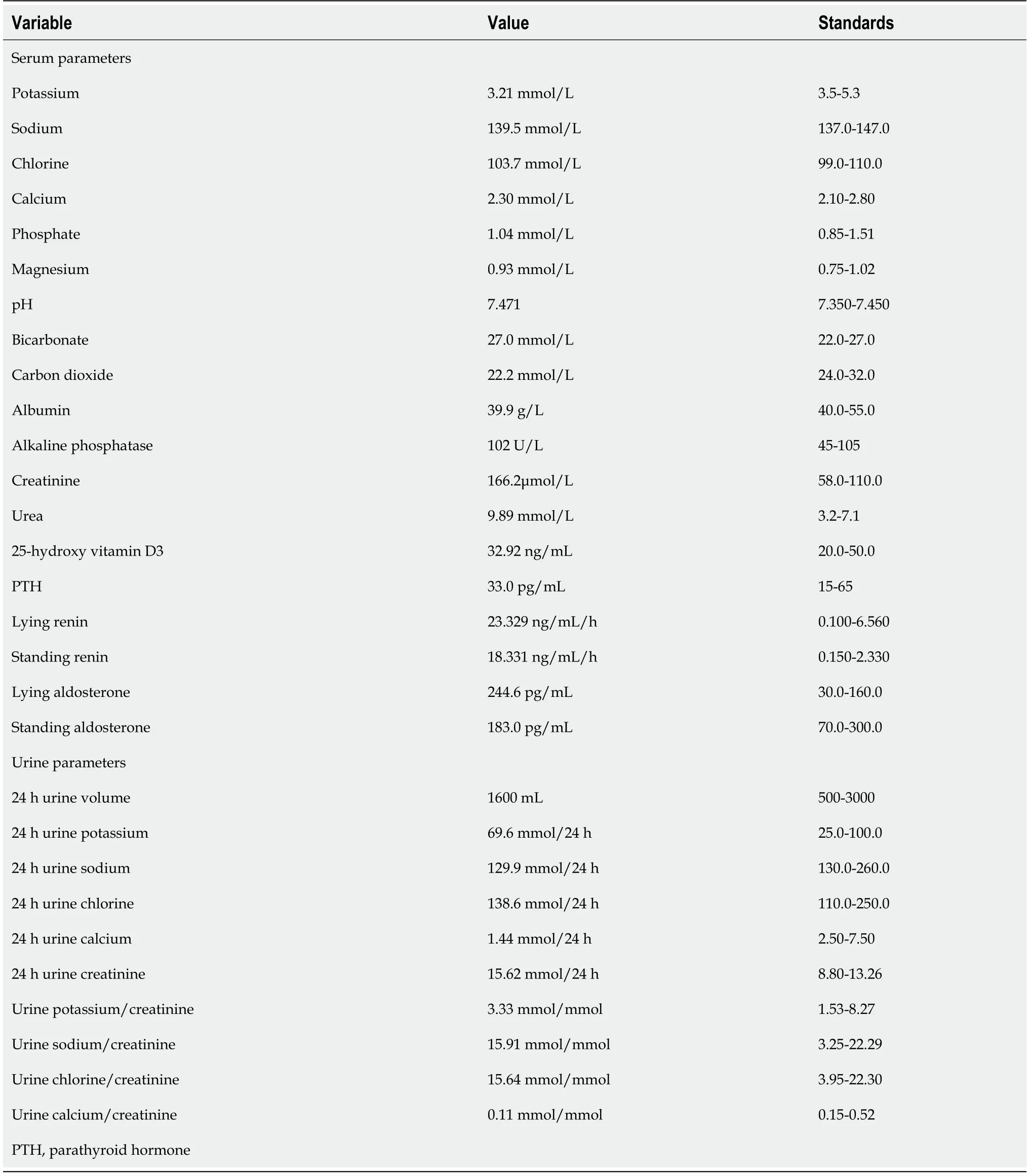
Table 1 Serum and urine biochemistry
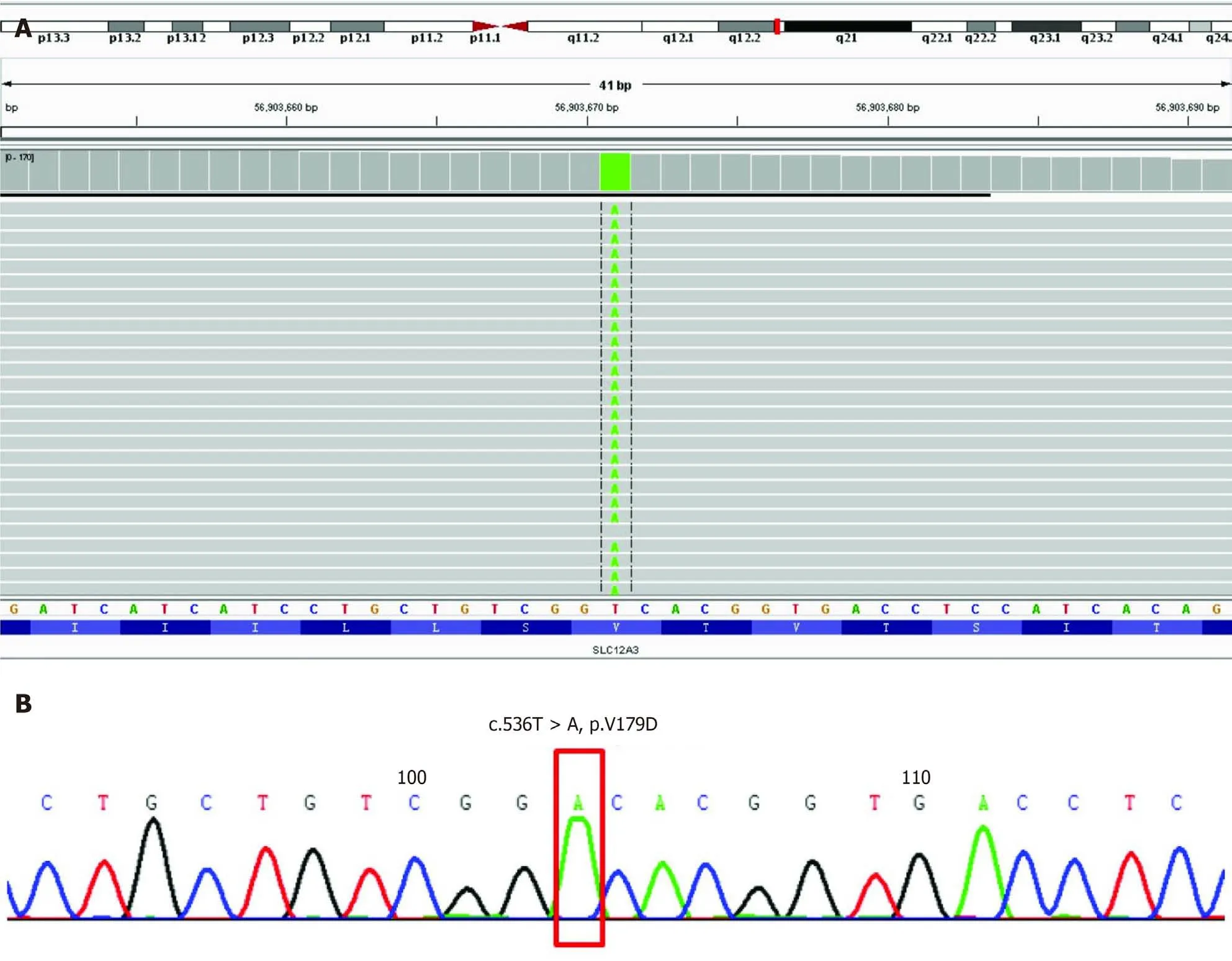
Figure 1 Genome sequencing of all exon regions of SLC12A3.A:Integrative Genomics Viewer of the genome sequencing revealed a homozygous mutation in C536T,which caused a Val to Asp substitution;B:The detected mutation is shown and was confirmed by classical Sanger sequencing.NM_000339.2 Solute Carrier Family 12 Member 3(SLC12A3):chr16:56903671;Exon4;c.536T>A;p.Val179Asp;homozygous mutation.
With the development of genetic testing technology,more cases of hypokalemia have been diagnosed as GS.The pathogenesis of GS is related to a mutation in theSLC12A3gene.At present,more than 400 mutation sites inSLC12A3have been found[9].These mutations include nonsense,missense,frameshift,deletion and insertion mutations.Heterozygous mutations are more common than homozygous mutations.Approximately 45% are compound heterozygous mutations occurring at two sites or more.However,the number of mutation sites showed no obvious correlation with clinical manifestations[4].TheSLC12A3gene encodes a thiazide diuretic-sensitive NaCl co-transporter consisting of 12 transmembrane domains with both the amino terminus and the carboxy terminus inside the cell[10].Mutations in theSLC12A3gene cause damage to the renal tubular Na+/Cl-transport function,resulting in a decrease in Na+and Cl-re-absorption in the distal collecting tube,leading to loss of NaCl,hypovolemia,hypotension,and metabolic alkalosis,followed by rising levels of renin and aldosterone.Increased reabsorption of Na+ through Na+ channels in epithelial cells on the cortical collecting duct is beneficial to the secretion of H+and K+[11].Abnormal Na+/Cl-transport can attenuate the hyperpolarization of Cl- inside the cells,increase Ca2+re-absorption,and reduce levels of urinary calcium as seen in our case[12].
The mechanism underlying GS hypomagnesemia is unclear.It may be related to the down-regulation of Mg2+M6 transient receptor potential channel protein on the apical membrane of distal convoluted tubules,resulting in increased renal magnesium excretion.The expression of M6 transient receptor potential channel protein in the intestine is down-regulated,the absorption of Mg2+in the intestine is reduced,and eventually the blood magnesium is decreased[13].Another reason for the decrease in blood Mg2+may be acceleration of Mg2+/Na+exchange on the luminal side caused by the increase in Na+re-absorption,which leads to increased magnesium in urine and decreased magnesium in blood[14].A total of 83.3% GS patients have hypomagnesemia,and only a few of have normal blood magnesium.GS patients with normal magnesium may make up a subtype accounting for 8% to 22% of cases reported in one study[15,16].Jianget al[14]observed 25 patients with GS and found that blood potassium in patients with normal magnesium(7/25)was slightly higher than those with hypomagnesemia(18/25).As hypomagnesemia can induce a further decrease in serum potassium,hypokalemia,hypochloremic metabolic alkalosis and RAAS activation are less severe in GS patients with normal magnesium than in GS patients with hypomagnesemia[15].
GS treatment mainly involves correcting disorders of electrolytes and metabolism,thereby alleviating clinical symptoms.For better efficacy,potassium supplementation should be combined with magnesium,aldosterone receptor antagonists,reninangiotensin system inhibitors(angiotensin-converting-enzyme inhibitors and angiotensin receptor blockers)and other drugs.Aldosterone receptor antagonists compound the renal salt wasting and should thus be started cautiously to avoid hypotension.Concomitant salt supplementation should be considered.Angiotensin converting enzyme inhibitors/angiotensin receptor blockers can also aggravate renal sodium wasting and increase the risk of symptomatic hypovolemia;these agents should be stopped in the case of acute,salt-losing complications,such as vomiting or diarrhea[1].It is very important to facilitate patient education which is an essential component of management.Patients are encouraged to eat more foods containing sodium chloride,potassium and magnesium as part of their diet.It is imperative that patients are aware of the adverse reactions of therapeutic drugs,especially gastrointestinal irritation caused by potassium chloride.In addition,there is a need to educate patients about what to do in the case of an emergency.Patients can exercise,but must pay special attention when participating in high-intensity and competitive sports,that can cause salt loss through sweating.During excessive sweating,diarrhea or vomiting,it is necessary to replenish electrolytes promptly to avoid serious complications[17].
Our patient had middle-aged GS onset,and the clinical manifestations were limb weakness and convulsions,but no hypertension.He had a family history of the condition.Laboratory examinations showed hypokalemia,normal blood magnesium level,metabolic alkalosis,hypocalciuria,proteinuria and RAAS activation.Through genome sequencing,the mutation c.536T > A(p.Val179Asp)ofSLC12A3gene was identified which supported our diagnosis of GS.This type of mutation is relatively rare.At present,few studies have reported cases of GS with proteinuria.Proteinuria might develop due to abnormalities of the glomerular basement membrane.Chronic kidney disease may develop in GS patients due to either chronic hypokalemia,which is associated with tubulointerstitial nephritis,tubule vacuolization,and cystic changes,or volume depletion and increased renin-angiotensin-aldosterone,which may contribute to renal damage and fibrosis.This patient also showed elevated creatinine,suggesting glomerular damage.He had hepatitis B virus infection;thus,we initially attributed the increase in creatinine to hepatitis B nephropathy.However,the possibility of other primary kidney diseases could not be eliminated.Unfortunately,the patient declined renal biopsy;therefore,the cause and progress of renal dysfunction required further investigation.Excluding other causes of renal impairment,and whether theSLC12A3gene mutation in GS patients affects podocyte function have not been reported and further supporting research is needed.
CONCLUSION
We report the case of a rare homozygous mutation in theSLC12A3gene in a Chinese patient with GS diagnosed by whole-exome sequencing,which showed that genome sequencing is a useful method for identifying genes involved in human genetic diseases.
ACKNOWLEDGEMENTS
We are grateful to the patient and his family for their willingness to participate in this study.
杂志排行

World Journal of Clinical Cases的其它文章
- Lymphoplasmacyte-rich meningioma with atypical cystic-solid feature:A case report
- Left bundle branch pacing with optimization of cardiac resynchronization treatment:A case report
- Arterial embolism caused by a peripherally inserted central catheter in a very premature infant:A case report and literature review
- Massive pulmonary haemorrhage due to severe trauma treated with repeated alveolar lavage combined with extracorporeal membrane oxygenation:A case report
- Successful treatment of encrusted cystitis:A case report and review of literature
- Mine disaster survivor’s pelvic floor hernia treated with laparoscopic surgery and a perineal approach:A case report