Primary pulmonary plasmacytoma accompanied by overlap syndrome: A case report and review of the literature
2020-04-08YiZhouXiaoHongWangShuangShuangMengHuiChaoWangYuXiaLiRuiXuXuHongLin
Yi Zhou, Xiao-Hong Wang, Shuang-Shuang Meng, Hui-Chao Wang, Yu-Xia Li, Rui Xu, Xu-Hong Lin
Yi Zhou, Department of Radiology, Huaihe Hospital of Henan University, Kaifeng 475000,Henan Province, China
Xiao-Hong Wang, Department of Invasive Technology, Huaihe Hospital of Henan University,Kaifeng 475000, Henan Province, China
Shuang-Shuang Meng, Yu-Xia Li, Rui Xu, Xu-Hong Lin, Department of Clinical Laboratory,Translational Medicine Center, Huaihe Hospital of Henan University, Kaifeng 475000, Henan Province, China
Hui-Chao Wang, Department of Nephrology, First Affiliated Hospital of Henan University,Kaifeng 475000, Henan Province, China
Abstract BACKGROUND Extramedullary plasmacytoma (EMP) is a rare kind of soft tissue plasma cell neoplasm without bone marrow involvement; this type of plasma cell neoplasm involves a lack of other systemic characteristics of multiple myeloma. Primary pulmonary plasmacytoma (PPP), with no specific clinical manifestations, is an exceedingly rare type of EMP. Because of its complexity, PPP is often difficult to diagnose, and there is no report in the literature on cases accompanied by overlap syndrome (OS).CASE SUMMARY A 61-year-old woman without a familial lung cancer history was admitted to our hospital in 2018, for intermittent cough, expectoration, and a stuffy feeling in the chest for 50 years; these symptoms appeared intermittently, especially occurred after being cold, and had been aggravated for the last 10 d. She was diagnosed with pulmonary fibrosis and emphysema, bronchiectasis, OS, and autoimmune hepatic cirrhosis in 2017. A pulmonary examination revealed rough breath sounds in both lungs; other physical examinations found no obvious abnormalities. A routine laboratory work-up showed decreased haemoglobin, increased ESR, and abnormal GGT, ALT, IgG, γ-globulin, κ-light chain, λ-light chain, rheumatoid factor, and autoimmune antibodies. Emission computed tomography demonstrated abnormally concentrated 99mTc-MDP. Chest computed tomography revealed a soft tissue mass in the middle and lower lobes of the right lung. After right middle and inferior lobe resection with complete mediastinal lymph node dissection, immunohistochemical analysis revealed an isolated pulmonary plasmacytoma. The patient received chemotherapy for more than 1.5 years and remains in good general condition.CONCLUSION PPP is a type of EMP, and we report an exceedingly rare presentation of PPP accompanied by OS.
Key Words: Pulmonary neoplasms; Plasmacytoma; Overlap syndrome; Case report
INTRODUCTION
Plasmacytoma is a type of malignant tumour derived from B lymphocytes.Plasmacytoma can be primarily classified into the following types: Multiple myeloma(MM), isolated plasmacytoma, extramedullary plasmacytoma (EMP), and plasmablastic sarcoma[1]. EMP is a monoclonal plasma cell tumour that originates from tissues other than the bone marrow haematopoietic tissue, and it represents 3%-5% of all plasmacytomas[2-4]. Wiltshaw[5]assumed that EMP can arise in almost any part of the body other than the bone marrow and haematopoietic tissues. Approximately 80%-90% of EMP cases involve the craniocervical structures (the nasal and paranasal cavities, nasopharynx, larynx, and upper aerodigestive tract); however, the number of cases is less than 1% of all neoplastic head and neck lesions[5-8]. The uncommon sites of EMP, to our knowledge, are the gastrointestinal and urogenital tracts, central nervous system, lymph nodes, and skin. Extremely rare cases have reported involvement of the lung[9,10].
Primary pulmonary plasmacytoma (PPP), with no specific clinical manifestations, is an exceedingly rare type of EMP. The diagnosis of PPP mainly depends on histopathological examination, while CD138 and CD38 are characteristically positive in plasmacytoma[11]. Overlap syndrome (OS) is generally defined as meeting criteria for more than one classic autoimmune connective tissue disease (CTD). Patients may present with evidence of more than one disease simultaneously, or they may develop different diseases sequentially. Some patients may exhibit two or more diseases with characteristic serologic markers for those conditions[12]. Herein, we present a typical presentation of PPP with OS.
CASE PRESENTATION
Chief complaints
A 61-year-old woman was referred to our hospital with complaints of intermittent cough, expectoration, and a stuffy feeling in the chest for the past 50 years, without chest pain, dyspnoea, fever, chills, sweating, nausea, vomiting, emesis, abdominal pain, weight loss, or loss of appetite. The above symptoms had been aggravated for the last 10 d.
History of present illness
The above symptoms appeared intermittently and always occurred after being cold during the past 50 years. She had no regular treatment in the past. She was diagnosed with OS (Sjogren’s syndrome and rheumatoid arthritis) three years prior and was mainly treated with glucocorticoids and immunosuppressants.
History of past illness
The patient had an appendectomy because of acute appendicitis approximately 30 years ago. She had swelling and pain of the bilateral knee, elbow, and wrist joints for more than 10 years, and the pain was intermittent. She also had manifestations of xerostomia, xerophthalmia, rampant dental caries, and chapped tongue for more than 10 years. She was diagnosed with Henoch-Schonlein purpura 10 years prior, and she was diagnosed with pulmonary fibrosis and emphysema, bronchiectasis, and autoimmune hepatic cirrhosis 3 years prior. She had parotid gland swelling when she was young, but she did not remember the details.
Personal and family history
The patient denied a history of smoking, tuberculosis, and alcohol or drug use. Her father died of oesophageal carcinoma, and her sister died of breast cancer. No family members had any similar clinical manifestations or death from similar diseases.
Physical examination
Her vital signs were as follows: Body temperature, 36.7°C; respiratory rate, 20 breaths/min; pulse rate, 79 bpm; and blood pressure, 17.5/10.1 kPa. Except for rough breath sounds without rales or rhonchi from both lungs, no obvious abnormalities were found in any other physical examinations.
Laboratory examinations
A routine laboratory work-up showed white blood cell count, 2.90 × 109/L; neutrophil percentage, 71.4%; haemoglobin, 78 g/L; ESR, 114 mm/h; C-reactive protein, 9.6 mg/L; GGT, 110 g/L; ALT, 300 U/L; total protein, 87.1 g/L; and albumin, 32.9 g/L.Serum calcium and phosphorus were within normal ranges. The rest of the routine laboratory examinations at the time of admission showed no obvious abnormalities.
Imaging examinations
A computed tomography (CT) scan of the chest revealed an irregular mass located in the middle and inferior lobe of the right lung (Figure 1A). The mass was nonhomogeneous and accompanied by spotted calcification, but without any area of necrosis on a CT plain scan (Figure 1B). It was lobulated and spiculated with adjacent pleural retraction and thus caused bronchial obstruction of the lateral segment of the right middle lobe and the right lower lobe. Enhanced scanning showed that the mass displayed moderate ununiform reinforcement with multiple tiny blood vessels(Figure 1C), and the pulmonary artery trunk was enlarged. Multiple enlarged lymph nodes were found in the mediastinum. A bone survey revealed no abnormalities.Echocardiography showed mildly reduced left ventricular function.
Further diagnostic work-up
The patient was further evaluated by sputum culture, and tests were negative. The full set of serum tumour markers, including CA-199 antigen (61.4 U/mL; normal range: ≤33 U/mL), and serum CA-125, CEA, NSE, and SCC were all in the normal range.Serum electrophoresis showed that albumin was 50.60% (normal range: 53.8%-65.2%)and γ-globulin was 29.20% (normal range: 9.2%-18.2%). Serum immunofixation electrophoresis revealed that IgG was 2520 mg/dL (normal range: 751-1560 mg/dL),κ-light chain was 2270 mg/dL (normal range: 629-1350 mg/dL), λ-light chain was 1260 mg/dL (normal range: 313-723 mg/dL), and monoclonal gammopathy was not found.The β2-microglobulin level was 6.07 µg/mL (normal range: 0.9-2.7 µg/mL). No Bence-Jones protein was detected in the patient’s urine.
Autoimmune examinations showed positivity for antinuclear antibody, anti-SSA/Ro60KD antibody, anti-SSA/Ro52KD antibody, anti-SSB antibody, and antineutrophil cytoplasmic antibody. The rheumatoid factor was 4077 IU/mL (normal range: 0-54 IU/mL), and the anti-cyclic citrullinated peptide antibody was 12.95 RU/mL (normal range: 0-25 RU/mL).
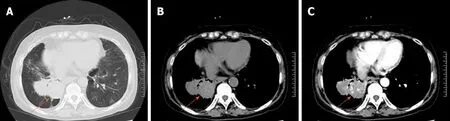
Figure 1 Computed tomography scan of the chest revealed an irregular mass (orange arrows) located in the middle and inferior lobe of right lung. A: Chest computed tomography image displayed at lung window showing a mass in the middle and inferior lobe of right lung, which caused bronchial obstruction of the lateral segment of the right middle lobe and the right lower lobe; B: Chest computed tomography image displayed at mediastinal window on a plain scan showing that the mass was lobulated, inhomogeneous, and accompanied by spotted calcification; C: Enhanced scanning in the artery phase showed that the mass displayed moderate ununiform reinforcement with multiple tiny blood vessels.
Emission computed tomography demonstrated that99mTc-MDP was abnormally concentrated at the bilateral eight front ribs, the right lower edge of the acetabulum,and the right inferior pubic ramus. A bronchoscopy was subsequently performed, and pathological examination showed that the bronchial mucosa of the right lower lobe was infiltrated with acute and chronic inflammatory cells, but no tumour cells were found. The bone marrow examination was unremarkable, with less than 5% of plasma cells and no dyscrasia.
FINAL DIAGNOSIS
The final diagnosis of the presented case was PPP accompanied by OS.
TREATMENT
The lesion of the right lung did not alleviate after anti-infection and hormone therapy,so the patient was originally treated by thoracoscopic right middle and inferior lobectomy. Unfortunately, her right middle and inferior lung lobes were atelectatic and solid; in addition, it was relatively difficult to separate the lesion from the surrounding chest wall and diaphragmatic muscle, the structure of the pulmonary hilum was unintelligible, and the fissure of the right lung was hypoplastic. Therefore,the surgery had to be converted to thoracotomy. Since the diagnosis from the frozen section was uncertain, we performed lymph node dissection. In addition, there was no pleural metastatic nodule.
Microscopic examination demonstrated the infiltration of numerous plasma cells,which coexisted with relatively mature and irregular immature plasma cells, and these cells were primarily basophilic. The nuclei were of different sizes, the nucleoli were large and often offset, and dual-core nucleoli and nuclear fission were observed.Immunohistochemical analysis of the tumour tissue showed LCA(+), CK(-), VIM(+),EMA(+), CD68(-), S-100(-), CD79a(+), CD3(-), Ki-67(+) (approximately 20%), CD38(+),CD138(+), κ(-), λ(-), and CD56(-). According to these results, the patient was eventually diagnosed with right lung plasmacytoma, and the tumour had not invaded the lymph nodes. Since ECT showed an abnormal concentration of99mTc-MDP, we suspected that the bone lesions were caused by plasmacytoma. After detailed and thorough communication, our patient decided to undergo postoperative chemotherapy.
OUTCOME AND FOLLOW-UP
The patient was treated with chemotherapy for more than 1.5 years after the surgery,and the short-term efficacy was acceptable. A chest CT scan demonstrated no new lesions or evidence of recurrence. At present, she is in good condition.
DISCUSSION
PPP is an exceedingly rare type of EMP[13]. PPP is generally divided into three stages based on the Wilshaw method[14]: Stage I, the tumour is confined to the primary site;stage II, the tumour has invaded local lymph nodes; and stage III, there are obvious widespread metastases. Therefore, in this case, the tumor should be classified as stage I.
A review[15]of 19 cases of PPP indicated that most patients with PPP were middleaged and elderly, with mean and median ages of 57 and 55 years, respectively. The male-to-female ratio was 1.4:1. Approximately 40% of patients ultimately developed MM, and the overall 2-year and 5-year survival rates were 66% and 40%, respectively.Josephet al[16]found that PPP is more prevalent in a younger group of patients (median age 43 years). The aetiology of the disease is not well understood, and we speculate that viral pathogenesis and chronic irritation are contributing factors[17]. Unfortunately,it presents non-specific manifestations and symptoms, and symptoms such as chronic cough, fever, expectoration, chest tightness, and haemoptysis have been reported. The clinical manifestation is closely related to lesion location. Compression or violation of the trachea and bronchi will cause dyspnoea, expectoration, and haemoptysis; the patient may experience chest pain if the pleura is involved[14]. PPP that presented with diffuse alveolar consolidation has been reported in published case reports[18-22], and these patients suffered from a pneumonia-like presentation. In this case, the patient presented with complaints of intermittent cough, expectoration, and a stuffy feeling in the chest after she had a cold. All of her symptoms could be attributed to pulmonary fibrosis and emphysema, bronchiectasis, and pulmonary infection.
Obviously, radiology plays an indispensable role in the diagnosis and differential diagnosis of PPP; unfortunately, the imaging features of PPP are non-specific. To differentiate PPP from MM, X-ray examination is necessary, and the patient should have a normal skeletal survey[23]. Typically, the radiographic presentation of PPP is a solitary mass or nodule, which is mostly localized to the lung hilar region but less commonly presents as lobar consolidation or diffuse pulmonary infiltrates[16]. Joseph[16]reported that PPP has a slight preference for the lower lobes, but other studies[9,15]demonstrated that both lungs are equally affected and that upper lobes are more likely to harbour lesions. Specifically, chest CT scans often show a single nodular,agglomerated soft tissue density, round or oval, with clear boundaries and uniform density. Necrosis and calcification are rarely seen in the lesion, but they can invade the hilar structure[24]. In a few cases[23], multiple pulmonary nodules or masses have been found. Some patients with a diffuse bilateral lung distribution can be revealed by lung consolidation through a CT scan, which has been reported in the literature[25]. PPP always shows moderate to significant enhancement after an enhanced chest CT scan,and abnormally increased vascular shadows are observed in and around the lesion[24,25].
Our patient's right lung had interlobular dysplasia, and the mass was lobulated and nonhomogeneous, with spotted calcification found on a CT plain scan. All of these findings were inconsistent with those previously reported in the literature[14,23-27]. The other difference is that the mass displayed moderate ununiform reinforcement on the enhanced CT scan. This patient was initially misdiagnosed with pulmonary sequestration, but after careful analysis, we found that there was no systemic blood circulation to the lesion. Therefore, the patient was misdiagnosed with lung cancer before the operation. Differential diagnoses of lymphoma, tuberculoma, bronchogenic carcinoma, and pulmonary segregation were considered but were eventually ruled out by characteristic findings from immunohistochemistry.
The diagnosis of PPP is complex, and definitive diagnosis requires histopathological examination of the specimen and immunohistochemistry. PPP microscopically shows dense plasma cells and different levels of diffuse proliferation and infiltration.Intranuclear inclusion bodies (Dutcher bodies) or eosinophilic inclusion bodies can be seen in tumour cells (PAS-positive inclusion bodies). Immunohistochemical staining reveals expression of a single light chain, with kappa lambda (−) or (+) or kappa lambda (+/−) predominating. Tumour cells are positive for CD138, CD38, CD45, PC,EMA, and CD20 but negative for CD15. In a few cases, CK and EMA are positive,while CD45 is negative. These features are in accordance with the typical histopathological characteristics of PPP. CD138 and CD38 are characteristically positive in plasmacytoma, especially CD138[14,18,28,29].
The diagnosis of PPP is made after rigorous investigation to rule out the presence of MM, and the diagnostic criteria for PPP are as follows[30,31]: Biopsy of tissue showing monoclonal plasma cell histology; bone marrow plasma cell infiltration not exceeding 5% of all nucleated cells; absence of osteolytic bone lesions or other tissue involvement(no evidence of myeloma); absence of hypercalcaemia or renal failure; and low serum M protein concentration, if present. In our case, the proportion of bone marrow puncture plasma cells was < 5%, suggesting that the plasma cells did not infiltrate the bone marrow. The pathological findings of the right lung lesion showed the infiltration of numerous plasma cells under the microscope, and immunohistochemistry suggested that the lung tumour cells were positive for CD138 and CD38 (Figure 2).The Bence-Jones protein was absent in her urine, and plasma electrophoresis for M protein and monoclonal gammopathy were negative. A skeletal survey showed no evidence of osteolytic lesions and no evidence of fracture. Thus, a diagnosis of PPP was made.
OS is defined as an entity that satisfies the classification criteria of at least two CTDs occurring at the same time or at different times in the same patient. OS is a kind of paraneoplastic syndrome, and the aetiology of the disease is not well known; it is speculated to be related to immune dysfunction[32]. Increasing evidence has demonstrated that autoantibodies are often involved in disease pathogenesis[12]. The characteristic of OS is that it often involves lung tissue, eventually causing pulmonary interstitial fibrosis[33]. Mixed connective tissue disease was the first OS that was defined in terms of association with a specific autoantibody[12]. A study[34]on the long-term outcomes of mixed connective tissue disease patients revealed that the majority of the patients died of pulmonary hypertension. In our case, the patient’s pulmonary artery trunk was enlarged, but there was no evidence to prove that she suffered from pulmonary heart disease, so we speculated that pulmonary hypertension may also be related to bland intimal proliferation and medial hypertrophy of the pulmonary arterioles. The treatment of OS is mainly based on the use of corticosteroids and immunosuppressive agents[35]. Unfortunately, we could not find a relationship between PPP and OS in the literature reported to date, but we speculated that the use of immunosuppressive agents may increase the risk of blood diseases, and we will explore this relationship further in future work.
On the basis of the literature, radiation therapy remains the standard treatment for PPP; other treatment modalities include resection alone or a combination of surgery with chemotherapy[16,20,30]. PPP consisting of a solitary lesion is usually treated by either surgical resection alone or resection followed by radiation therapy. PPP is a special kind of EMP; when small in size and detected in its early stages, it is usually sensitive to radiotherapy, which leads to complete remission in the majority of cases[1,30,36-38].However, when pulmonary involvement is diffuse, chemotherapy is the best choice[18-21]. Chemotherapy is indicated for patients with refractory and/or relapsed disease; melphalan and prednisone are commonly used[19,20,23], and the present case also responded well to chemotherapy with melphalan and prednisone. Adjuvant chemotherapy should be considered in patients with tumours larger than 5 cm and those with high-grade tumours[39]. Compared to those of other plasma cell neoplasms,a favourable prognosis has been reported for EMP. With such treatments, recurrence and dissemination rates between 20% and 40% were observed and ten-year survival is typically 70%[1,7]. Patients with PPP can have long-term survival, as evidenced by Koss’s[15]finding that two of these patients survived 20 or more years.
In this case, our patient underwent surgical resection with complete resolution of the tumour, combined with chemotherapy after the surgery. For several reasons, such as financial circumstances for her family, she eventually received a vincristine,doxorubicin, and dexamethasone regimen for four cycles, followed by bortezomib along with dexamethasone for two cycles. During the treatment with vincristine,doxorubicin, and dexamethasone regimens, adverse reactions, such as bone marrow suppression and drug-induced hepatitis, appeared; fortunately, the patient’s general condition was acceptable after symptomatic treatment. After communicating with the patient, we changed the chemotherapy regimen. She has undergone six cycles of chemotherapy to date. Thoracic CT was reviewed several times after the surgery, and no suspected finding of disease recurrence/progression had been found up to January 14, 2020.
CONCLUSION
In summary, PPP, a rare primary pulmonary malignant tumour, is relatively difficult to diagnose because of its non-specific clinical manifestation. We present a case of PPP accompanied by OS. This report suggests that when physicians find blood-rich lung nodules or masses, they should take into account the possibility of PPP. A thorough evaluation is essential to exclude systemic disease, and further close clinical follow-up should be performed for a relatively long time.

Figure 2 Histopathological examinations. A: The pathological findings of the right lung lesion showed the infiltration of numerous plasma cells under the microscope, the nuclei were in different sizes, and the nucleoli were large and often offset (Hematine-eosin staining, × 40); B: Immunohistochemistry suggested that the right lung tumour cells were positive for CD138, which was specific to plasmacytomas; C: The right lung tumour cells were positive for CD38 on immunohistochemistry staining.
ACKNOWLEDGEMENTS
We would like to thank every member of our team, with special thanks to Dr. Lin for her careful guidance during the process of writing this article.
杂志排行

World Journal of Clinical Cases的其它文章
- Relationship between non-alcoholic fatty liver disease and coronary heart disease
- Remission of hepatotoxicity in chronic pulmonary aspergillosis patients after lowering trough concentration of voriconazole
- Endoscopic submucosal dissection as alternative to surgery for complicated gastric heterotopic pancreas
- Observation of the effects of three methods for reducing perineal swelling in children with developmental hip dislocation
- Predictive value of serum cystatin C for risk of mortality in severe and critically ill patients with COVID-19
- Sleep quality of patients with postoperative glioma at home