Idiopathic multicentric Castleman disease with pulmonary and cutaneous lesions treated with tocilizumab: A case report
2020-04-08PingYangHanHuiHuiChiYuTongSu
Ping-Yang Han, Hui-Hui Chi, Yu-Tong Su
Ping-Yang Han, Hui-Hui Chi, Yu-Tong Su, Department of Rheumatology and Immunology, Ruijin Hospital, Shanghai Jiao Tong University School of Medicine, Shanghai 200025, China
Ping-Yang Han, Department of Rheumatology and Immunology, the Affiliated Hospital of Hangzhou Normal University, Hangzhou 310015, Zhejiang Province, China
Abstract BACKGROUND Human herpes virus-8 (HHV-8)-negative, idiopathic multicentric Castleman disease (iMCD) is a rare and life-threatening disorder driven by proinflammatory cytokines, which is still poorly understood. Pulmonary parenchyma lesion is a rare condition in iMCD, which mainly manifests as lymphocytic interstitial pneumonia and is an indicator of severe iMCD. Cutaneous lesion is also very rare and mainly occurs in Asians. There have been few reports of iMCD patients with both skin and lung parenchyma involvement.CASE SUMMARY We present a Chinese man who complained about a 3-year history of intermittent dry cough and a 2-year history of diffuse reddish-brown maculopapules.Laboratory examination revealed polyclonal hypergammaglobulinemia and hypercytokinemia including interleukin 6. Chest computed tomography revealed small patchy shadows with ground-glass nodules scattered in two lobes and mediastinal lymphadenopathy. The pathological result of the lymph node was consistent with the plasma cell type of Castleman disease. As serum human immunodeficiency virus test and HHV-8 staining of the lymph node were negative, the patient was finally diagnosed with HHV-8 negative iMCD. He was treated with tocilizumab at an intravenous (i.v.) dose of 8 mg/kg every 2 wk combined with methylprednisolone at an i.v. dose of 80 mg/d initially with gradual dose tapering. Partial remission was achieved 9 mo later.CONCLUSION iMCD with lung parenchyma and skin involvement is a rare condition that requires clinicians’ attention and awareness for early diagnosis.
Key Words: Multicentric Castleman disease; Lymphocytic interstitial pneumonia; Skin involvement; Interleukin 6; Tocilizumab; Case report
INTRODUCTION
Castleman disease (CD) describes a group of poorly understood lymphoproliferative disorders driven by proinflammatory hypercytokinemia. It can be classified as unicentric (UCD) or multicentric (MCD) according to the lesions involved. Some cases of MCD are caused by human herpes virus-8 (HHV-8) whereas others are HHV-8-negative and defined as idiopathic MCD (iMCD)[1]. Liu AYet al[2]reported that HHV-8-negative iMCD is relatively rare, accounted for about one-third of all published cases of MCD, and the onset age of iMCD is between 2- and 80-years-old (median: 50-yearsold) with male predominance. It is more common in Asian populations, especially Japanese, with 6.25% of all cases occurring in China. Although the etiology of iMCD remains unclear, its pathogenesis is currently thought to be mainly related to abnormal secretion of the cytokine interleukin 6 (IL-6)[3]. Patients have heterogeneous clinical features, characteristic lymph node histopathology, and often deadly multiple organ dysfunction. Anti-IL-6 monoclonal antibodies (mAbs) combined with/without glucocorticoids therapy had been recommended as the first-line treatment regimen for iMCD[4]. Both skin and lung parenchyma involvement were rare in iMCD. Herein, we report a case of patient with pulmonary involvement as the primary manifestation accompanied with peculiar multiple skin eruptions, who was finally diagnosed with iMCD. Treatments with anti-IL-6 mAb combined with glucocorticoids were effective.
CASE PRESENTATION
Chief complaints
A 32-year-old Chinese man was admitted to our hospital complaining about a 3-year history of intermittent dry cough and a 2-year history of diffuse reddish-brown maculopapules. In the meantime, he experienced a gradual increase in physical weakness and malaise over the past 2 mo.
History of present illness
The patient developed a recurrent dry cough 3 years ago, which broke out in day and night. However, he denied obvious sputum, fever, shortness of breath, or joint pain.He had been treated with several kinds of empirical antibiotics with no improvement.Two years ago, a gradually increased scattered reddish and brown maculopapules began to appear on his face, neck, and trunk without itch, ecdysis or blisters. There were no symptoms on limbs. Although the patient had visited several dermatologists,the diagnosis remained vague, and effective treatment was not obtained. Two months ago, he began to feel a gradually progressed physical weakness and malaise.
History of past illness
The patient was born, raised, and has been living in Shanghai. He was allergic to sulfonamides and aspirin. He underwent appendectomy because of appendicitis 2 years ago. Otherwise, he denied infections, bad habits, or any family history.
Physical examination
On physical examination, he was not feverish with oxygen saturation 98% in room air.Innumerable symmetric brownish-red macules, papules and plaques about 0.2-1 centimeters in diameter were distributed on his face, neck, chest, abdomen and back(Figure 1A). Besides, there were palpable lymph nodes without tenderness in cervical,axillary and inguinal regions.
Laboratory examinations
On laboratory tests, he had normocytic anemia with a hemoglobin of 11.6 mg/dL(reference range 13.1-17.2 mg/dL) and thrombocytosis with a platelet count of 341 ×109/L (reference range 85-303 × 109/L). Total serum protein, erythrocyte sedimentation rate (ESR), and C-reactive protein (CRP) elevated to 102 g/L (reference range 65-85 g/L), 112 mm/h (reference range 0-15 mm/h), and 91.6 mg/L (reference range < 10 mg/L) respectively, while serum albumin decreased to 23 g/L (reference range 35-55 g/L). Polyclonal immunoglobulin (Ig) elevated with IgG 5350 mg/dL (reference range 751-1560 mg/dL), IgA 572 mg/dL (reference range 82-453 mg/dL), IgE 430.0 IU/mL(reference range 5.0-165.3 IU/mL), and IgG4 1590 mg/dL (reference range 3-200 mg/dL). Blood and urine immunofixation electrophoresis showed no abnormal monoclonal bands. Serum cytokine assay revealed the levels of multiple cytokines elevated with vascular endothelial growth factor (VEGF) 780.42 pg/mL (reference range 0-142 pg/mL), IL-6 39.3 pg/mL (reference range < 3.4 pg/mL), IL-8 75.6 pg/mL(reference range < 62 pg/mL), soluble IL-2 receptor 1692.0 U/mL (reference range 223-710 U/mL) and tumor necrosis factor > 24.1 pg/mL (reference range < 8.1 pg/mL).Other laboratory values including tests for infections and other immunological parameters were unremarkable.
Imaging examinations
The chest computed tomography (CT) conducted 3 years ago revealed that small patchy shadows with ground-glass nodules scattered in bilateral lobes with mediastinal lymphadenopathy. The chest CT has been repeated several times in the recent 2 years, which showed gradually progressed pulmonary lesions accompanied with new developed cysts in upper lobes, and lymphadenopathy developed in the mediastinum, hilum, and bilateral axillary fossa (Figure 1C). Ultrasonography revealed that the largest lymph node was located in the right groin with the diameter of 2.2 cm × 1.2 cm. Positron emission tomography-CT showed no neoplastic lesions except for enlarged lymph nodes in multiple sites, hepatosplenomegaly, and diffuse lesions of bilateral pulmonary parenchyma. Pulmonary function was basically normal.Fiberoptic bronchoscopy found no new organisms or bleeding, and bronchoalveolar lavage fluid analysis revealed no evidence of infection or malignancy.
Pathological examinations
A thoracotomy biopsy of the middle lobe of right lung revealed fibrous tissue hyperplasia accompanied by infiltration of a few lymphocytes and plasma cells(Figure 2A and B). The immunohistochemistry demonstrated that the infiltrated plasma cells were positive for both κ and λ chains without light chain restriction. Bone marrow aspiration and biopsy found no evidence of neoplastic or preneoplastic diseases.
Further diagnostic work-up
Furthermore, biopsies of the abdominal papules and the right inguinal lymph node by surgical resection were performed. Histopathology of the skin lesions showed that the epidermis was basically normal; lymphoid follicular-like hyperplasia was observed in the superficial and deep layers of the dermis, and medium-density plasma cells and lymphocytes infiltrated around the blood vessels and glands with no atypia (Figure 2C and D). Histopathology of the lymph node showed that lymphoid reactive hyperplasia mainly occurred in the paracortical region, and plasma cells infiltrated mainly in the medulla region; the structure of the lymph node was intact, but no transparent small angiogenesis was found (Figure 2E and F). Immunohistochemistry (IHC) of the skin and lymph node revealed that the number of IgG, IgM, κ and λ staining-positive plasma cells were comparable, with few IgG4-positive plasma cells. There was no light chain restriction. The test of Epstein–Barr virus (EBV)in situhybridization and HHV-8 by the IHC staining of latency-associated nuclear antigen 1 in the lymph node specimen was negative. Several experienced pathologists in our hospital analyzed the histopathology of the lung, skin, and lymph node, a diagnosis of the plasma cell type of CD was finally considered.
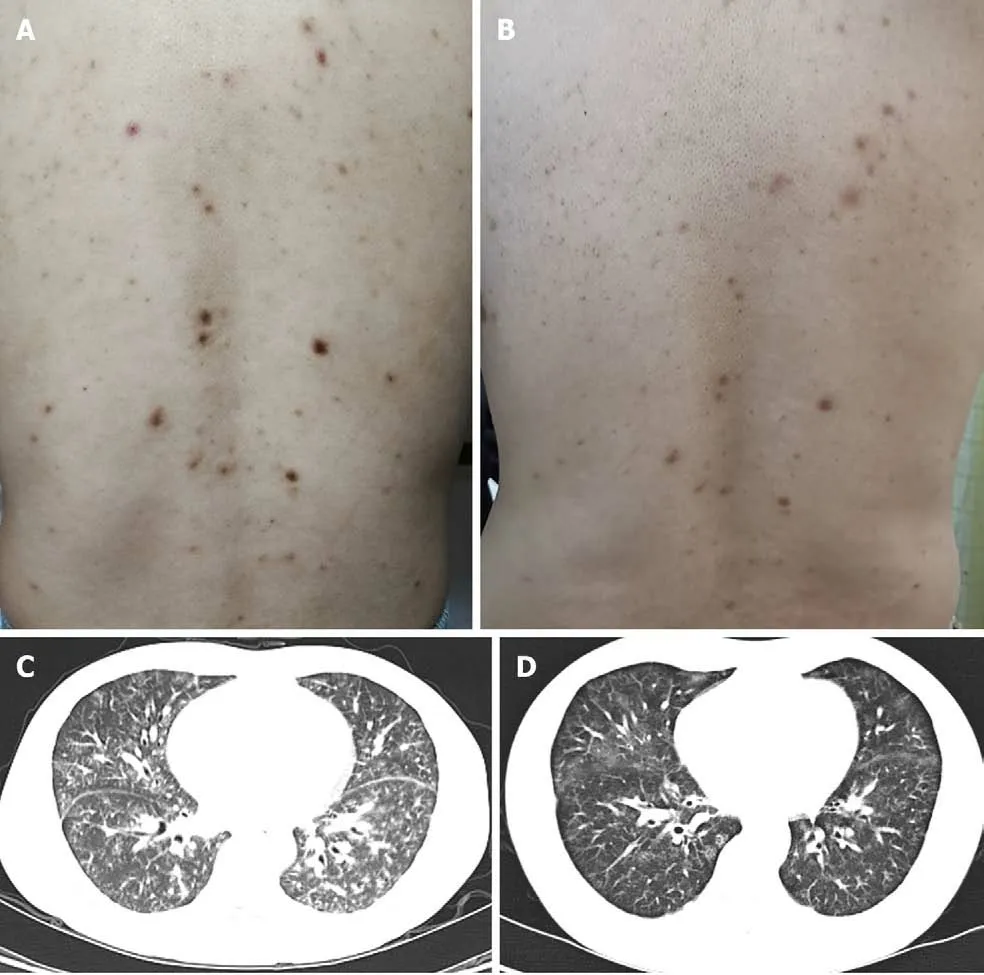
Figure 1 Skin lesions and chest computed tomography findings before and after treatment. A: Innumerable symmetric brownish-red macules,papules, and plaques about 0.2-1 cm in diameter distributed in the back before treatment; B: The skin lesion improved 9 mo after treatment; C: Parenchymal window setting of the chest computed tomography showed diffuse ground-glass opacities in two lobes before treatment; D: The lung lesion improved 9 mo after treatment.
FINAL DIAGNOSIS
The final diagnosis of the presented case was iMCD.
TREATMENT
According to the iMCD treatment recommendation published in 2018[4], as siltuximab is not available in China, the patient was treated with tocilizumab at an i.v. dose of 8 mg/kg every 2 wk combined with methylprednisolone at an i.v. dose of 80 mg/d initially with dose tapered gradually. The patient tolerated the treatment well and did not complain apparently adverse reactions except a mild skin itch.
OUTCOME AND FOLLOW-UP
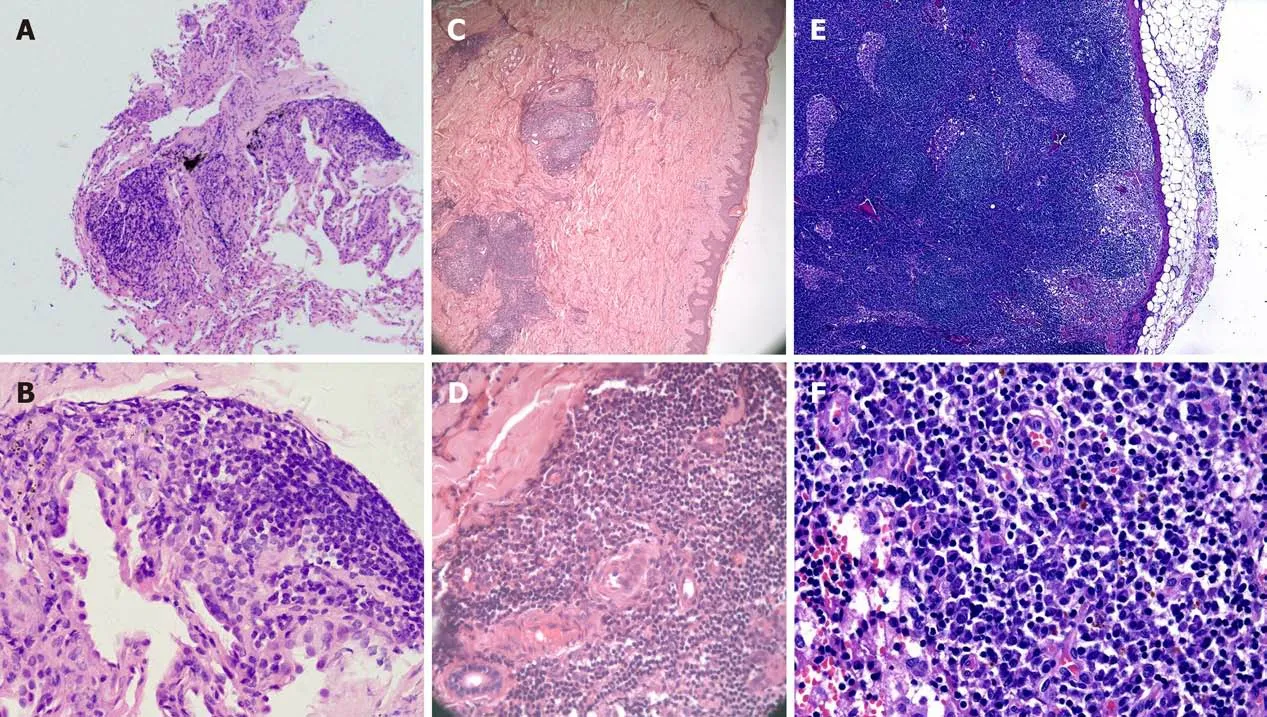
Figure 2 Histopathology of the lung (A and B), skin (C and D), and lymph node (E and F). A, C, and E: Hematoxylin and eosin (H&E) × 50; B, D, and F: H&E × 400. A and B: Fibrous tissue hyperplasia accompanied by infiltration of a few lymphocytes and plasma cells in the lung; C and D: The epidermis was basically normal, lymphoid follicular-like hyperplasia was observed in the superficial and deep layers of the dermis, and medium-density plasma cells and lymphocytes infiltrated around the blood vessels and glands with no atypia; E and F: Reactive lymphoid hyperplasia mainly occurred in the paracortical region, and plasma cells mainly infiltrated in the medulla region. The structure of the lymph node was intact, and no transparent small angiogenesis was found.
Nine months later, the patient’s constitutional symptoms were significantly relieved.Meanwhile, the levels of serum albumin, hemoglobin, and platelet count returned to normal ranges, and the elevated levels of ESR, CRP, Ig, and cytokines were partially reduced. The cutaneous and lung lesions improved apparently (Figure 1B and D).
DISCUSSION
HHV-8-negative iMCD was a rare disease, and the clinical manifestations are heterogeneous, which mainly presented as fever, night sweats, weight loss, ascites,pleural effusions, lymphadenopathy, hepatosplenomegaly, and polyclonal hypergammaglobulinemia[2,5]. It was unwonted to see both skin and lung parenchyma involvement in iMCD. Here, we report a case of patient with pulmonary and skin involvement as the primary manifestation who was finally diagnosed with iMCD. The patient had presented to several tertiary hospitals for more than 3 years before he was finally diagnosed. According to the iMCD diagnostic criteria developed in 2017[1], our patient met both two main criteria (histopathological features consistent with iMCD on an excisional lymph node biopsy and lymph nodes larger than 1 cm in short-axis diameter in two or more lymph node station), three clinical minor criteria(constitutional symptoms, hepatosplenomegaly, and lymphocytic interstitial pneumonitis [LIP]), and five laboratory minor criteria (elevated CRP and ESR, anemia,thrombocytosis, hypoalbuminemia, and polyclonal hypergammaglobulinemia). In addition, infections such as HHV-8, HIV, and EBV, autoimmune diseases such as systemic lupus erythematosus and rheumatoid arthritis, and malignancies such as lymphoma were excluded. Moreover, he had hypercytokinemia including IL-6 and VEGF, so that he was finally diagnosed with iMCD.
Although approximately 70% of CD cases occurred in the thorax, solitary mass (s)and/or mediastinal and/or hilar lymphadenopathy were the most common radiological manifestations for both UCD and MCD[6,7]. MCD-associated diffuse parenchymal lung disease was uncommon, and LIP-like findings were the typically reported radiological imaging pattern[8-11]. In addition to LIP-like performance, diffuse pulmonary parenchyma can also be expressed as multiple nodules of different size and sites, patchy, ground-glass opacities or consolidation in their chest CT[12,13]. Most patients tend to appear obvious lymphadenopathy in the hilum and/or mediastinum,and some patients showed mass in the mediastinum or hilum[11-13]. Cough, fever, and difficulty in breath were the main clinical manifestations with them. LIP is a clinicopathologic entity in the spectrum of benign pulmonary lymphoproliferative disorders, and most of these patients have a polyclonal hypergammaglobulinemia[14].Thin-walled cysts in random distribution are the characteristic abnormality, along with other imaging features of ground-glass opacities, poorly defined centrilobular and subpleural nodules, reticulonodular opacities, and alveolar consolidation with air bronchograms and thickening of the interstitial along the lymphatic vessels[8,14]. The presence of cysts may help to differentiate LIP from lymphoma.
Cutaneous involvement similar to that noted in the lymph nodes is also extremely rare in CD. Such skin involvement in CD might have been overlooked or unreported,because CD is not well recognized in the dermatological field. Skin involvement of MCD is often reported as cases, and the skin lesions often manifest as that resembled cutaneous and systemic plasmacytosis (CSP)[15-18], or eruptive cherry hemangiomatosis or violaceous papules[19-22], and most of these patients are Asians. At present, CSP is considered a variant of iMCD, and CSP and cutaneous iMCD are considered as the same disease entity[15,16,23]. CSP is a rare disorder characterized by an infiltration of mature plasma cells in various organ systems. The cutaneous morphology is characterized by red to dark brown macules, papules and plaques a few centimeters in diameter, usually distributed symmetrically on the face, neck and back. To date, CSP has primarily been presented as of case reports and no more than 100 cases have been reported. Most cases are in Asians and almost exclusively in Japanese[23].
There are few reports of CD patients with both skin and lung parenchyma involvement, which are mainly reported in cases of CSP[24-26]. Miyagawa-Hayashino Aet al[25]reported a 54-year-old Japanese man with lymphadenopathy and interstitial lung disease diagnosed with CSP. The skin lesions manifested as diffuse red-brown papules on his trunk and face, and chest CT revealed that ground-glass opacities and slight reticular shadows in the middle and lower lobe bilaterally. Histopathology of the skin showed dense perivascular and periadnexal infiltration of plasma cells in the deep dermis, and histologic findings of the lung showed thickened alveolar septa due to the lymphoplasmacytic infiltration. This is similar to our patient’s symptom,although the chest CT showed that the lung lesion is less severe compared to our patient.
The diagnosis of iMCD requires a pathological confirmation of an excisional lymph node biopsy. MCD histopathological features can be divided into four variants:hyaline-vascular, plasma cell, mixed, and plasmablastic. Histopathological features of patients with iMCD showed an enrichment of the plasma cell and mixed pathology variants, with relatively fewer cases reporting hyaline vascular changes[2,5]. The histopathology of the lymph node in our patient was consistent with the plasma cell type of MCD. Moreover, the histopathology of the skin, lung and bone marrow of the patients showed an infiltration of polyclonal plasma cells, which indicated multi-organ involvement.
According to the guideline, monoclonal antibodies (mAbs) targeting IL-6 directly(siltuximab) or the IL-6 receptor (tocilizumab) have been approved for iMCD therapy.Siltuximab (11mg/kg every 3 wk) is recommended (category 1) for all patients with non-severe iMCD, and it is presently approved in the United States, Canada, Europe,and Brazil, among other countries. If siltuximab is not available, tocilizumab (8 mg/kg every 2 wk) may be used (category 2A), and it is approved for the treatment of iMCD in Japan. The availability of siltuximab and tocilizumab varies amongst countries, and the choice between the two drugs is currently more dependent on indication within that country and access, as no head-to head trials have been performed to compare efficacy[4]. Since siltuximab is not available in China, our patient received a treatment of tocilizumab combined with glucocorticoid, his clinical symptoms and laboratory indicators were improved apparently. For iMCD, relapses occur on cessation of therapy, an indefinite continuation of anti-IL-6 mAb therapy in responding patients is therefore recommended[4]. Meanwhile the incidence of malignancy in patients with iMCD is increased[2], the patient still requires a long-term consistent follow-up and evaluation from us.
In summary, iMCD is a rare and multisystem lymphoproliferative disease. The frequent delayed diagnosis still remains a concern. The literature reports that the average delayed diagnosis time of iMCD is 3 mo[5]. We need to improve the recognition of iMCD with pulmonary symptoms as the initial presentation for early and timely diagnosis and treatment. Besides, the diagnosis also requires the assistance from experienced pathologists. In clinical practice, for patients with lung parenchyma and skin involvement with polyclonal hypergammaglobulinemia as the initial manifestations, the physicians should be aware of the diagnosis of iMCD. The cytokine assessment, biopsies and multidisciplinary consultation can help to confirm the diagnosis.
CONCLUSION
iMCD with lung parenchyma and skin features as the primary manifestations is a rear clinical condition. Polyclonal hypergammaglobulinemia and lymphadenopathy are the two major clinical features of iMCD. Diagnosis requires the confirmation of histopathology of a lymph node by excision in combination with other clinical pictures. Anti-IL-6 mAbs combined with glucocorticoid is an effective frontline treatment regimen, especially for those with an elevated level of serological IL-6. As the rate of malignancy in patients with iMCD is increased comparing to the general population, and LIP can progress to acute respiratory failure without an appropriate therapy, a long-term follow-up with an appropriate therapy is desperately needed.
ACKNOWLEDGEMENTS
Innovative research team of high-level local universities in Shanghai.
杂志排行

World Journal of Clinical Cases的其它文章
- Relationship between non-alcoholic fatty liver disease and coronary heart disease
- Remission of hepatotoxicity in chronic pulmonary aspergillosis patients after lowering trough concentration of voriconazole
- Endoscopic submucosal dissection as alternative to surgery for complicated gastric heterotopic pancreas
- Observation of the effects of three methods for reducing perineal swelling in children with developmental hip dislocation
- Predictive value of serum cystatin C for risk of mortality in severe and critically ill patients with COVID-19
- Sleep quality of patients with postoperative glioma at home