基于生物信息学分析筛选舌鳞状细胞癌核心基因及其预后价值
2020-04-07黄成易尚辉查文婷吕媛
黄成 易尚辉 查文婷 吕媛


摘要:目的 通過对GEO数据库提供的基因芯片数据进行挖掘,结合生物信息学分析基因表达谱,获取舌鳞状细胞癌(TSCC)核心基因,利用生存分析初步验证核心基因对舌鳞状细胞癌的预测效果。方法 从GEO数据库下载舌鳞状细胞癌相关芯片数据(GSE9844),获得了26例TSCC组织样本和12例癌旁组织样本的全基因组转录组谱,采用SAM算法筛选出TSCC与癌旁组织间的差异表达基因,并借助GEO的gene信息库对基因功能进行描述,筛选出TSCC与癌旁组织间的差异细胞信号通路,构建决定TSCC的基因共表达网络,通过GEPIA数据库来初步验证共表达网络中的核心基因是否与TSCC患者的生存预后存在相关性。结果 筛选出2074个差异表达基因,包括1119个上调基因和955个下调基因。以2074个差异表达基因作为共表达网络的构建基础,共纳入230个差异表达基因,筛选出5个TSCC核心的基因(ADCY4、PLA2G12A、MAOB、PDE2A、CYP2C9),通过GEPIA数据库对核心基因进行生存分析,初步验证共表达网络中高表达的ADCY4基因与TSCC总体生存率呈正相关(P=0.014),高表达PLA2G12A基因与TSCC总体生存率呈负相关(P=0.0029),MAOB、PDE2A及CYP2C9基因患者生存率比较,差异无统计学意义(P>0.05)。结论 通过生物信息学方法分析影响TSCC的核心基因,最终筛选出2个差异表达非常显著且对患者预后影响明显的基因,对TSCC的诊断和预后治疗提供了新思路,提高TSCC机制的研究效率。
关键词:舌鳞状细胞癌;生物信息学;核心基因;共表达网络
中图分类号:R739.91 文献标识码:A DOI:10.3969/j.issn.1006-1959.2020.03.002
文章编号:1006-1959(2020)03-0006-07
Screening Core Genes of Tongue Squamous Cell Carcinoma Based
on Bioinformatics Analysis and Its Prognostic Value
HUANG Cheng,YI Shang-hui,ZHA Wen-ting,LYU Yuan
(Hunan Normal University Medical College,Changsha 410001,Hunan,China)
Abstract:Objective To obtain core genes of tongue squamous cell carcinoma (TSCC) by mining gene chip data provided by the GEO database and analyzing gene expression profiles in combination with bioinformatics, and use survival analysis to initially verify the role of core genes in tongue squamous cell carcinoma forecast effect. Methods Tongue squamous cell carcinoma-related chip data (GSE9844) was downloaded from the GEO database, and the genome-wide transcriptome profiles of 26 TSCC tissue samples and 12 adjacent cancer tissue samples were obtained. The differences between TSCC and adjacent cancer tissues were screened using the SAM algorithm express genes, and use GEO's gene information database to describe gene functions, screen differential signaling pathways between TSCC and adjacent tissues, construct a gene co-expression network that determines TSCC, and use the GEPIA database to initially verify the co-expression network. Whether the core genes are correlated with the survival prognosis of patients with TSCC.Results 2074 differentially expressed genes were screened, including 1119 up-regulated genes and 955 down-regulated genes. Using 2,074 differentially expressed genes as the basis for the construction of a co-expression network, a total of 230 differentially expressed genes were included, and 5 TSCC core genes (ADCY4, PLA2G12A, MAOB, PDE2A, CYP2C9) were selected, and the core genes were survived through the GEPIA database. The analysis showed that the highly expressed ADCY4 gene in the co-expression network was positively correlated with the overall survival rate of TSCC (P = 0.014), the highly expressed PLA2G12A gene was negatively correlated with the overall survival rate of TSCC (P = 0.0029), and patients with MAOB, PDE2A, and CYP2C9 genes,there was no significant difference in survival rate (P> 0.05).Conclusion The core genes affecting TSCC were analyzed by bioinformatics methods. 2 genes with very significant differential expression and significant effects on patients' prognosis were finally screened, which provided new ideas for the diagnosis and prognosis of TSCC and improved the research efficiency of TSCC mechanism.
Key words:Tongue squamous cell carcinoma;Bioinformatics;Core genes;Co-expression network
生物信息学是20世纪80年代末随着人类基因组计划的启动而兴起的一门交叉学科,体现了生物学、计算机科学、数学、物理学等学科间的渗透与融合。其通过对生物学实验数据的获取、加工、存储、检索与分析,达到揭示数据所蕴含的生物学意义从而解读生物活动规律的目的,在后期的临床试验、新药研制以及复杂疾病早期诊断、治疗中具有重要指导意义[1-5]。舌鳞状细胞癌(tongue squamous cell carcinoma,TSCC)作為头颈部最为常见的癌症,具有恶性化程度高、浸润生长速度快、易转移等特点,可累及舌肌导致吞咽、言语功能受损[6],目前的流行病学研究显示[7],TSCC发病率逐年上升,并且有年轻化的趋势。虽然目前的临床治疗手段不断提升,但是TSCC容易淋巴转移的特点导致其总体生存率不高,TSCC的侵袭和淋巴转移已经成为其预后不佳的主要原因[8]。舌癌的发生是多基因参与、多步骤发生的生物学过程,包括多基因的功能失常,如原癌基因激活和抑癌基因失活等,目前对TSCC的生物信息学研究较多,但大多筛选出的基因仅停留在实验室研究阶段,应用于临床实践的相关报道罕见。TSCC的发生与发展是一个多基因及相关因子作用的结果,随着高通量测序和基因芯片技术的不断发展,为TSCC的预防和治疗,以及发现导致TSCC的核心基因提供了基础保障[9]。本研究利用GEO数据库中TSCC基因芯片数据筛选出TSCC差异表达基因,对差异基因进行生物信息学分析及共表达网络构建,以期获取TSCC 的核心基因及相关通路[10]。通过GEPIA数据库对核心基因进行生存分析,初步验证核心基因的表达与TSCC的关系,为进一步研究TSCC发生发展的作用机制提供科学依据。
1资料与方法
1.1数据资料收集 本研究中所使用的基因芯片表达谱来自GEO数据库(网址:https://www.ncbi.nlm.nih.gov/geo/),所有患者的表达谱数据均来自相同的实验平台(Affyme-trix U133 Plus 2.0 array)。
1.2差异基因筛选方法 本研究在筛选差异基因阶段所采用的SAM(significance analysis of microarrays)算法[11]是一种专业针对DNA芯片数据进行分析的算法,由美国Standford大学开发,其本质为一种高通量的生物信息分析平台。采用GEO 数据库自带的GEO2R分析实验室分析GSE9844中TSCC样本及癌旁组织中的差异表达基因,差异基因筛选标准:Fold change>1.2且P<0.05。
1.3细胞信号通路分析方法 GO功能富集分析一般包括生物过程(biological process,BP)、分子功能(molecular function,MF)和细胞成分(cellular component,CC),本研究中对基因的生物过程、分子功能和细胞成分进行富集分析。将这些差异表达基因导入到在线工具DAVID 数据库中,分别进行GO 和KEGG富集分析,P<0.05表示差异有统计学意义[12]。利用KEGG数据库来识别下调差异表达基因以及上调差异表达基因中显著富集的通路。通路数据资源均来源自kyoto encyclopedia of genes and genomes database(KEGG) 数据库。KEGG数据库(http://www.genome.ad.jp/kegg)是一组数据库与基因信息相关联在一起的软件,用于通过其基因组信息帮助理解与模拟细胞或者生物系统中的高阶功能行为[13-17]。
1.4基因共表达网络分析方法 利用在线数据库String构建差异表达基因之间的交互作用网络图(PPI),分析蛋白质功能之间的相互作用。随后我们应用 Cytoscape 软件(3.5.1)在线工具使 PPI 的网络模块可视化,使用 Cyto Hubba 插件对差异基因共表达网络进行关联度分析,并根据节点度(degree)大小进行排序,筛选出核心基因[18,19]。
1.5生存分析 通过GEPIA数据库对TSCC核心基因进行在线生存分析,筛选条件为LUAD数据集,95%置信区间,时间轴单位为月。基因表达差异采用t检验,在TSCC中表达量与预后的关系采用Log-rank检验,以P<0.05表示差异有统计学意义。
2结果
2.1TSCC基因芯片样本类型分布 本研究从美国国立生物技术信息中心共收集基因芯片样本38例,TSCC样本26例(68.42%);癌旁组织基因芯片样本12例(31.58%)。
2.2差异表达基因筛选结果 本研究共获得2074个差异表达基因,火山图中红色为上调基因(见图1),蓝色为下调基因。其中包括1119个上调基因和955个下调基因,q-value为误判率,即差异基因中假阳性基因所占比例的期望,值越小假阳性率越低。图2为2074个差异表达基因的聚类热图。在样本聚类中,红色代表TSCC组织样本,绿色代表正常癌旁组织样本。在基因聚类中,红色代表基因表达水平较高,绿色代表基因表达水平较低。表1为DEGs列表(局部),TSCC组织中基因表达水平与正常癌旁组织相比较,MMP1基因在癌组织上调基因中差异倍数最高,TMPRSS11B在下调基因中差异倍数最高。
2.3细胞信号通路富集分析结果 针对2074个差异表达基因的细胞信号通路富集结果显示,富集度最高的信号通路为ECM-receptor interaction,富集度为8.63,富集度排名前5的信号通路还包括Metabolic pathways通路、PI3K-Akt signaling pathway、Focal adhesion通路及Pathways in cancer通路。前20位差异细胞信号通路富集结果比较,差异均有统计学意义(P<0.001),可认为本研究得到的信号通路富集结果具有较高的真实性和有效性,见表2、图3。
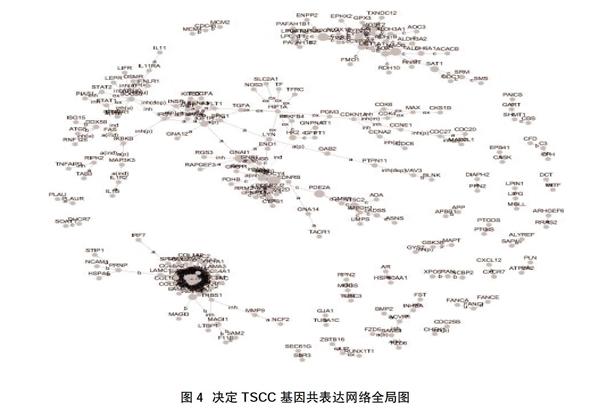
2.4基因共表达网络结果 以2074个在不同组发生差异表达的基因作为共表达网络的构建基础,本次构建得到的共表达网络共纳入230个发生差异表达的基因。以TSCC(TSCC)为实验组,以正常癌旁组织为对照组,采用MCODE算法计算基因调控能力评分的结果显示,决定TSCC调控能力评分最高基因为ADCY4基因,得分为425.2分,该基因在本次构建得到的模块中上游基因数为10,下游基因数为6,这在某种程度上说明ADCY4基因在决定TSCC的过程中虽具有较强的调控能力,但其功能的发挥在很大程度上受到其上游基因的影响。
此外,PLA2G12A、MAOB、PDE2A以及CYP2C9基因的调控能力评分均高于或接近于200,因此可认为它们是本次构建得到的共表达网络模块中的核心基因。本次构建得到的共表达网络模块中所有纳入基因的调控能力评分以及上下游基因数参见表3,决定TSCC基因共表达网络全局图见图4。
2.5决定TSCC的共表达网络基因功能描述 通过TSCC基因共表达网络MCODE算法得分,筛选出得分排在前二十位的基因,其中排名在前五位的基因分别是:ADCY4(腺苷酸环化酶4)、PLA2G12A(磷脂酶A2,XIIA组)、MAOB(单胺氧化酶B)、PDE2A(磷酸二酯酶2A,cGMP刺激)和CYP2C9(细胞色素P450,家族2,家族C,多肽9),见表4。
2.6 TSCC中5种核心基因与患者预后关系 PLA2G12A表达水平对患者的总生存时间有着显著影响(P<0.05)。而ADCY4对患者生存率也有明显影响(P<0.05),MAOB、PDE2A以及CYP2C9对患者的生存率影响无统计学意义(P>0.05),见图5。
衡量富集度的大小。因此,细胞信号通路的富集度越大,就越能认为该信号通路在疾病的发生发展中起着重要的作用[23]。
对TSCC差异基因进行网络化分析,以基因之间的关系为线,实线表示激活作用,虚线表示抑制作用,如果在共表达网络中不知道某个基因的功能,可以通过调控的相关节点基因和周围的连线来判断该基因的功能。此处经常用到基因调控能力评分来对基因在共表达网络中的调控能力进行量化评估,其计算原理主要涉及 MCODE 代码算法,其本质为一种寻找共表达网络局部稠密区域的算法,找到网络图的局部密集地区后,MCODE 代碼基于聚类系数,使用顶点加权方案。MCODE代码算法包括三个阶段(顶点加权、复杂预测和可选后处理),其原理类似于聚类分析(都是通过指标的降维进而选取有代表性的指标),但有别于聚类分析的是,共表达网络在构建的过程中放大了重要关联图区域的权重,因此在进行核心基因筛选的过程中具有一定的算法优势[24]。
以上的研究表明通过生物信息学方法来寻找TSCC的生物标志物和其对临床预后可能有重要意义,通过相关的文献来进一步证明这些核心基因在舌癌和其他肿瘤的发生发展中起到重要的作用[25,26],以期为对舌癌的诊断和预后治疗提供了新思路,提高舌癌机制的研究效率,为将来对TSCC的早期诊断和基因靶向治疗提供了科学依据和理论指导。
参考文献:
[1]Chan LL,Jiang P.Bioinformatics analysis of circulating cell-free DNA sequencing data[J].Clin Biochem,2015,48(15):962-975.
[2]Ranganathan S,Tan T,Schonbach C.InCoB2014:bioinformatics to tackle the datato knowledge challenge.Introduction[J].BMC Bioinformatics,2014,15(16):1471-2105.
[3]Ow TJ,Upadhyay K,Belbin TJ,et al.Bioinformatics in otolaryngology research.Part one: concepts in DNA sequencing and gene expression analysis[J].J Laryngol Otol,2014,128(10):848-858.
[4]Meldolesi E,van Soest J,Damiani A,et al.Standardized data collection to build prediction models in oncology:a prototype for rectal cancer[J].Future Oncol,2016,12(1):119-136.
[5]Perry PM.Harnessing the power of big data and data analysis to improve healthcare entities[J]. Healthc Financ Manage,2016,70(1):74-75.
[6]D?觟brossy L.Epidemiology of head and neck cancer:Magnitude of the problem[J].Cancer Metastasis Rev,2005,24(1):9-17.
[7]Timar J,Csuka O,Remenar E,et al.Progression ofhead and neck squamous cell cancer[J].Cancer Metastasis Rev,2005,24(1):107-127.
[8]Mackenzie J,Ah-See K,Thakker N,et al.Increasing incidence of oral cancer amongst youngpersons:what is the aetiology[J].Oral Oncol,2000,36(4):387-389.
[9]Annertz K,Anderson H,Biorklund A,et al.Incidence and survival of squamouscell carcinoma of the tongue in Scandinavia,with special reference to young adults[J].Int J Cancer,2002,101(1):95-99.
[10]Ye H,Yu T,Temam S,et al.Transcriptomic dissection of tongue squamous cell carcinoma[J]. BMC Genomics,2008,9(1):69-70.
[11]Jez S,Martin M,South S,et al.Variants of unknown significance on chromosomal microarray analysis:parental perspectives[J].J Community Genet,2015,6(4):343-349.
[12]Kanehisa M,Sato Y,Morishima K.BlastKOALA and GhostKOALA:KEGG Tools for Functional Characterization of Genome and Metagenome Sequences[J].J Mol Biol,2016,428(4):726-731.
[13]Cheng L,Lin H,Hu Y,et al.Gene function prediction based on the Gene Ontology hierarchical structure[J].PLoS One,2014,9(9):e84685.
[14]Kanehisa M,Sato Y,Kawashima M,et al.KEGG as a reference resource for gene and protein annotation[J].Nucleic Acids Res,2016,44(D1):17.
[15]Blake JA,Chan J,Kishore R,et al.Gene Ontology Consortium:going forward[J].Nucleic Acids Research,2015,43(Database issue):1049-1056.
[16]Nigrovic PA,Muscal E,Riebschleger M,et al.AMIGO:a novel approach to the mentorship gap in pediatric rheumatology[J].J Pediatr,2013,164(2):226-227.e1-e3.
[17]Peltola MA,Kuja-Panula J,Liuhanen J,et al.AMIGO-Kv2.1 Potassium Channel Complex Is Associated With Schizophrenia-Related Phenotypes[J].Schizophr Bull,2016,42(1):191-201.
[18]Zhou T,Zhang Y,Wu P,et al.Potential biomarkers and latent pathways for vasculitis based on latent pathway identification analysis[J].Int J Rheum Dis,2014,17(6):671-678.
[19]魏選东.基于芯片分析的乳腺癌预后核心基因筛选及其预测效果分析[D].湖南师范大学,2018.
[20]Iancu OD,Colville A,Darakjian P,et al.Coexpression and cosplicing network app roaches for the study of mammalian brain transcriptomes[J].Int Rev Neurobiol,2014,116(1):73-93.
[21]汪涛,蒋庆华,彭佳杰,等.基因共表达网络的构建及分析方法研究综述[J].智能计算机与应用,2014(6):51-54,57.
[22]洪胜君.基于转录组测序数据的基因共表达网络研究[D].复旦大学,2013.
[23]王安训.舌鳞状细胞癌侵袭和转移的研究进展[J].口腔疾病防治,2016,24(5):261-266.
[24]Nsman A,Bersani C,Lindquist D,et al.Human papillomavirus and po-tentiallyrelevant biomarkers in tonsillar and base of tongue squamouscell carcinoma[J].Anticancer Res,2017,37(10):5319-5328.
[25]高桂林,朱斌,颜孟雄.舌鳞状细胞癌相关差异基因的生物信息学及预后分析[J].临床口腔医学杂志,2018(3):145-149.
[26]Langfelder P,Horvath S.WGCNA:an R package for weighted correlation network analysis[J]. BMC Bioinformatics,2008,9(1):559.
收稿日期:2019-12-24;修回日期:2020-01-10
编辑/肖婷婷
