“非保护型”金属胶体纳米簇形成机理研究
2020-04-02陈丽芳于聿律桑雅子程涛刘岩村上洋原田雅史王远
陈丽芳,于聿律,桑雅子,程涛,刘岩,村上洋,原田雅史,*,王远,*
1北京大学化学与分子工程学院,分子动态与稳态结构国家重点实验室,北京分子科学国家研究中心,北京 100871
2 Department of Health Science and Clothing Environment, Faculty of Human Life and Environment, Nara Women’s University,Nara 630-8506, Japan.
3 Institute for Quantum Life Science, National Institutes for Quantum and Radiological Science and Technology (QST), Umemidai 8-1-7, Kizugawa-city, Kyoto 619-0215, Japan.
1 引言
金属纳米簇是尺寸小、分布窄的金属纳米粒子,其独特的表面效应和电子性质使其对许多化学反应具有优异的催化性能1-7。金属纳米簇表面原子处于高度配位不饱和状态,具有较高的表面自由能,因此在制备金属纳米簇胶体溶液时常需要加入高分子、表面活性剂和强有机配体等保护剂,利用空间位阻等作用实现金属纳米簇的稳定化。然而,在很多情况下,这些保护剂的存在不利于金属纳米簇功能体系的组装和金属纳米簇本征性质的研究8。
“非保护型”金属纳米簇由表面吸附的溶剂分子和简单离子实现稳定化,它们可以作为构筑复相催化剂的结构基元。将预先制备的非保护型金属或合金纳米簇负载于不同的载体表面,可以区分复相催化剂中的尺寸、组成、载体表面基团以及修饰剂对催化性质的影响;合成结构可控的纳米复合物;制备兼具高金属载量和高金属分散度的复相催化剂,它们是构筑燃料电池催化电极等功能体系的关键材料。
迄今,小尺寸、窄分布的金属纳米簇的制备方法尚十分有限。我们曾发明了一种“非保护型”金属纳米簇及其合成方法(碱-乙二醇法)9,该方法可以高效合成Pt、Rh、Ru、Ir、Os等一系列非保护型金属及合金纳米簇。此类金属纳米簇由表面吸附的乙二醇分子和简单离子实现稳定化,具有尺寸小、分布窄、稳定性高、制备效率高、易于分离和修饰的特点,可以与许多有机或无机材料复合,制备性能优异的纳米复合催化剂。此类金属纳米簇及其合成方法已被广泛用于研制新型高效催化剂10-18和燃料电池催化电极19-33。
我们将碱-乙二醇法制得的非保护型Ru纳米簇负载于氧化锡载体上,制备了纳米复合催化剂Ru/SnO2。Ru/SnO2在催化卤代硝基苯加氢反应中表现出比Ru纳米簇胶体更高的催化活性和选择性,催化反应速率达到4.3 × 10-2mol·mol-1·s-1,SnO2与Ru纳米簇的相互作用有效地抑制了其催化氯代硝基苯氢化反应中C―Cl键的氢解,产物中氯代苯胺的选择性大于99.9%10。将非保护型Pt纳米簇与氧化铁载体复合,制备了对卤代硝基苯氢化制备卤代苯胺反应具有高催化活性和高选择性的催化剂。γ-Fe2O3和Fe3O4负载的Pt纳米簇是目前对上述催化反应性能最好的催化剂。X光子能谱、化学吸附和CO红外探针实验结果表明,Pt纳米簇向氧化铁表面氧空穴转移电子,这可能是此类催化剂在上述反应中表现出高选择性的重要原因11,12。
我们将非保护型Pt纳米簇和Ru纳米粒子沉积于Fe3O4载体上,制备了Fe3O4负载的Ru和Pt双金属催化剂,该催化剂可以在313 K下催化CO2和H2反应生成多碳醇和多碳烃(C2-C6)13。同位素标记实验结果证明该催化体系中高碳醇由催化剂表面金属烷基键的水解生成,这一新的催化转化途径不同于以往基于CO插入烷基金属键的高温途径,使低温催化CO2氢化转化为高碳醇得以实现。研究表明Fe3O4负载的Ru纳米粒子可以在低温下有效催化CO2加氢生成CHx*物种,而Fe3O4负载的Pt纳米粒子则可以有效催化C―C偶联和烷基水解反应生成多碳醇,催化剂中Pt和Ru纳米粒子的协同作用使得该催化剂可以在温和条件下催化CO2转化为多碳醇和多碳烃。Zeng等人14使用非保护型Ru纳米簇与SiO2纳米线组装制得纳米复合催化剂Ru/SiO2,该催化剂在350 °C下可高选择性地催化CO2加氢生成CO,催化剂的活性达到0.04 s-1。
Somorjai等人15,16利用上述非保护型Pt纳米簇等催化材料,系统研究了乙烯加氢催化剂中Pt纳米粒子尺寸对催化活性的影响。Kunz等人17使用α-氨基酸修饰碱-乙二醇法制备的非保护型Pt纳米簇,研究了金属纳米簇表面功能配体与底物相互作用对反应物手性诱导的影响,为调控不对称加氢反应产物的立体选择性提供了有益的思路。
碱-乙二醇法制备的非保护型金属及合金纳米簇,在能源转化与利用、电化学催化剂的研制中得到了广泛的应用19-33。Xin19-21、Wan22、Mao23等人在利用非保护型Pt纳米簇发展新型燃料电池催化剂方面开展了先驱性的研究工作。近年来,我们将非保护型PtCu合金纳米簇与蜜勒胺修饰的碳载体(MMC)复合,合成了PtCu/MMC催化剂,其在电催化氧还原反应(ORR)中表现出优异的催化性能24。其中,Pt75Cu25/MMC在0.9 V处的质量活性高达1.59 A·mg-1(每毫克贵金属,下同),比活性为3.98 mA·cm-2(每平方厘米铂),是目前已报道的ORR催化活性最高的PtCu合金纳米簇基催化剂。与之前所报道的PtCu电催化剂25相比较,Pt75Cu25/MMC具有更高的稳定性,经过10000次循环老化测试后,其质量活性仅下降9%。实验数据清楚地揭示了MMC载体显著增强了PtCu合金纳米簇对ORR的催化活性和催化剂的稳定性。使用非保护型PtRu合金纳米簇与氮掺杂碳纳米角(NCNHs)复合制备的PtRu/NCNHS纳米复合催化剂在电催化甲醇氧化反应(MOR)中表现出良好的活性,其质量活性为850 mA·mg-126。
Liu等人27合成了一系列具有不同原子比的非保护型PtPd合金纳米簇并将其负载于介孔碳(OMC)载体上,所制备的Pt3Pd/OMC催化氧还原反应的比活性和质量活性分别是商用Pt/C (Pt: 20%(w))催化剂的2.5倍和4.1倍。Lee等人28使用非保护型Pt纳米簇与碳纳米管(CNT)复合制备的Pt/CNT催化剂在ORR中表现出较高的稳定性。
作为构筑催化剂的结构基元,碱-乙二醇法制备的非保护型金属纳米簇已在催化研究领域中得到了广泛的应用。然而,对形成此类非保护型金属纳米簇详细机理的认识有待进一步提高。普遍接受的形成过程为氯化物前驱体与碱反应形成氢氧化物或氧化物纳米粒子,后者被乙二醇还原生成金属纳米簇9,这种成核与还原分步进行的策略使此类金属纳米簇的尺寸受底物浓度的影响较小。Liu等人34利用XPS分析了碱-乙二醇法合成Pt纳米簇过程中间物种的价态,在中间物种中同时测到了Pt(IV)、Pt(II)和Pt(0)三种价态,作者认为这一结果支持了碱-乙二醇法的两步反应机理。另一方面,Hwang及其合作者35,36利用X射线吸收精细结构谱(XAFS)分析了碱-乙二醇法制备PtRu合金纳米簇的形成过程,作者认为在反应过程中,前驱体先被还原为而RuCl3与NaOH反应生成PtRu合金纳米簇是由乙二醇还原和得到的。
近年来,时间分辨的原位实验技术得到了快速发展,使液相中金属纳米簇形成机理的深入研究成为可能。Harada等人37利用原位快速X射线吸收光谱(QXAFS)对H2PtCl6在乙醇溶液中的光还原过程进行分析。结果表明,在光照还原H2PtCl6的过程中,Pt(IV)先被还原形成中间物种Pt(II),后者再被还原至金属态Pt(0)。而在纳米粒子的形成过程中,存在还原-成核过程(reduction-nucleation)、表面自催化-生长过程(autocatalytic surface growth on nucleates)和奥氏熟化(Ostwald ripening-based growth)三种过程。Norbert Steinfeldt38利用原位小角散射(SAXS)技术研究了碱-乙二醇法中n(NaOH)/n(H2PtCl6)之比对纳米粒子尺寸的影响。
在本项研究中,我们利用原位QXAFS和原位紫外-可见(UV-Vis)吸收光谱研究了碱-乙二醇合成法中金属纳米簇的形成过程与机理;同时利用TEM和DLS技术分析了反应中间物种的尺寸变化。结果表明,在上述非保护型Pt胶体纳米簇的形成过程中,部分Pt(IV)在NaOH乙二醇溶液中被还原为Pt(II),Pt(IV)、Pt(II)与OH-反应的产物进一步缩合生成氧化物纳米粒子,最终氧化物纳米粒子被还原至Pt金属纳米簇。在碱-乙二醇法合成非保护型Ru纳米簇的过程中,RuCl3与OH-反应的产物进一步缩合形成氧化物纳米粒子,后者被还原形成Ru金属纳米簇。
2 实验部分
2.1 试剂
水合氯铂酸(H2PtCl6·6H2O,分析纯)和水合三氯化钌(RuCl3·xH2O,优级纯)购买于国药集团化学试剂公司,氢氧化钠(NaOH)和乙二醇(EG)均为分析纯,购买于西陇化工股份有限公司。
2.2 原位实验
将RuCl3/EG或H2PtCl6/EG前驱体溶液与NaOH/EG溶液预先在原位测试池外部混合,再由蠕动泵注入原位反应池,随后在加热反应物的过程中进行原位测试。原位反应测试系统如图1所示,其中原位反应池的光路长度为2 mm。
非保护型Pt胶体纳米簇([Pt]: 40 mmol·L-1)的具体制备过程如下:
(1)将H2PtCl6溶解于乙二醇,并定容至 50 mL容量瓶,配制成浓度为80 mmol·L-1的H2PtCl6/EG溶液。
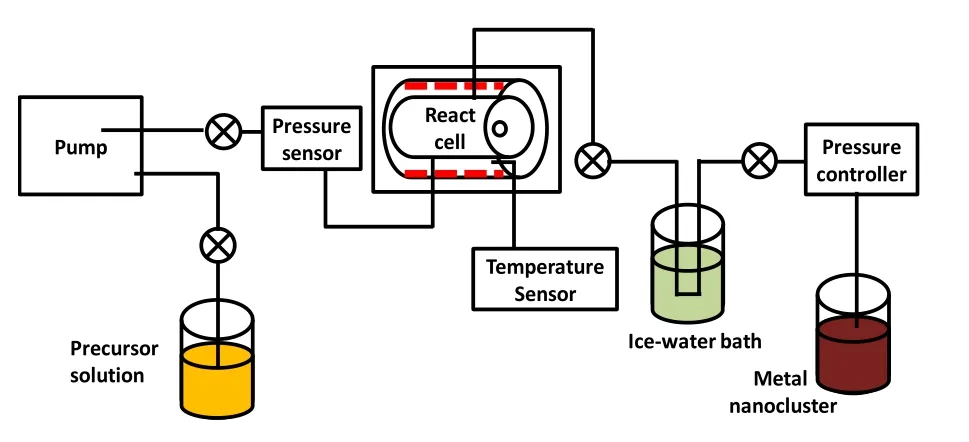
图1 原位反应池系统示意图Fig.1 Schematic graph of in situ reaction cell system.
(2)在搅拌下将50 mL 0.53 mol·L-1NaOH的乙二醇溶液逐滴加入到步骤(1)中配置的H2PtCl6乙二醇溶液中,继续搅拌15 min,得到橙黄色混合溶液。
(3)将上述混合溶液泵入原位反应池,关闭原位反应池入口和出口处的阀门,然后开启加热控温装置,使原位反应池的温度从室温升到80 °C并在80 °C保持2 h。反应过程中每分钟采集反应体系的QXAFS和UV-Vis吸收光谱。
非保护型Ru胶体纳米簇([Ru]:40 mmol·L-1)的具体制备过程如下:
(1)将RuCl3溶解于乙二醇,并定容至50 mL容量瓶中,配制成浓度为80 mmol·L-1的RuCl3/EG溶液。
(2)搅拌下将50 mL 0.26 mol·L-1NaOH/EG溶液逐滴加入到步骤(1)中配置的RuCl3/EG溶液中,搅拌15 min,得到深棕色混合溶液。
(3)将上述混合溶液泵入原位反应池,关闭原位反应池入口和出口处的阀门,然后开启加热控温装置,使原位反应池的温度从室温升到100 °C,并在100 °C保持3 h。反应过程中每分钟采集反应体系的QXAFS和UV-Vis吸收光谱。
2.3 测试与表征
非保护型Pt纳米簇形成过程中的原位QXAFS测试和参比样品的EXAFS测试均是在日本高能加速器研究机构(KEK,PF)的BL-9C束线上进行的。该束线的电子储存环能量为2.5 GeV,环形电流最大值设置为450 mA。调节电离室的检测气组成使X射线的辐射强度具有最佳值,其中入射强度(I0)的电离室检测气为Ar(15%)N2(85%)混合气体,透射强度(I)的电离室检测气为纯Ar。以两块平行的Si(111)单晶作为能量单色器,在QXAFS测试过程中,能量单色器连续快速地移动,使入射X射线的能量快速变化(几秒到几分钟),从而保证原位测试实验的信号采集。每个光谱采集吸收边前(-230 eV)到吸收边后(+995 eV)能量范围内的吸收强度,共采集约2000个数据点,获得一组完整吸收光谱的用时设置为1 min。
非保护型Ru纳米簇形成过程中的原位QXAFS测试和参比样品的EXAFS测试则是在日本高能加速器研究机构(KEK,PF-AR)的NW-10A束线上进行。该束线的电子储存环能量为6.5 GeV,环形电流最大值设置为55 mA。以两块平行的Si(311)单晶作为该束线的能量单色器,用于电离室检测束线强度的气氛分别为纯Ar气体(I0)和纯Kr气体(I)。其它测试条件与非保护型Pt纳米簇的条件相同。
透射电镜照片(TEM)和高分辨透射电镜照片(HRTEM)的拍摄分别是在场发射电子显微镜FEI Tecnai G2F20 (操作电压为200 kV)和FEI Tecnai G2F30 (操作电压为300 kV)上进行。电镜样品的具体制备过程如下:采用油浴加热反应混合溶液,设置Pt体系的反应温度为80 °C,Ru体系的反应温度为100 °C,使反应体系从室温升到设定温度并维持该温度反应2-3 h,反应过程中取样。
动态光散射(DLS)测试是在Brookhaven 90Plus/BI-MAS纳米/亚微米激光粒度分析仪上进行。光散射的样品制备过程同电镜样品的制备,取适量液体样品稀释到合适浓度,置于洁净的石英池中,平行测试5组。
3 结果与讨论
3.1 非保护型Pt胶体纳米簇的形成机理
不同金属离子在UV-Vis吸收光谱中具有特定吸收峰,且其吸收强度与金属离子浓度间存在良好的线性关系,因此,紫外-可见分光光度法是检测金属胶体纳米簇制备过程中各反应物种浓度变化的有效方法之一。
图2a为EG、NaOH/EG溶液、H2PtCl6/EG前驱体溶液及H2PtCl6/EG和NaOH/EG混合溶液的UVVis吸收光谱。从图中可以观察到,乙二醇溶剂和NaOH/EG溶液在测试波长范围内没有吸收峰,而H2PtCl6/EG溶液的吸收光谱中有两个特征吸收峰,其中强度较大的吸收峰位于364 nm左右,另一个强度较弱的吸收峰位于464 nm左右。室温下向H2PtCl6/EG溶液中加入过量的NaOH (NaOH/Pt摩尔比 = 6.6),其吸收光谱没有明显变化。
图2b为制备非保护型Pt胶体纳米簇反应体系的原位UV-Vis吸收光谱。从图中可以观察到,当温度升至78.3 °C时,反应体系光谱中已有明显的纳米粒子的散射信号,说明此时反应体系中已生成可被UV-Vis吸收光谱检测到的纳米粒子。当混合溶液在80 °C下反应超过30 min,UV-Vis吸收光谱的散射信号强度基本不在变化,意味着反应体系中纳米粒子的形成及生长过程已基本完成。
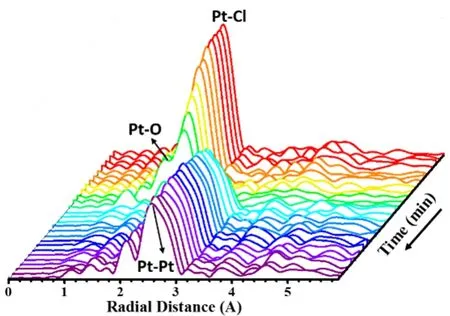
我们利用原位XAFS技术分析了非保护型Pt金属纳米簇的形成过程,图3为PtL3边的X射线吸收近边结构(XANES)。PtL3边的XANES谱中,吸收边后的第一个吸收峰称为白线峰(White Line,简称WL),对应于电子由Pt 2p3/2能级向费米能级附近未占据的Pt 5d能级的跃迁。白线峰的吸收强度反映样品中Pt的5d能级电子填充状态。白线峰强度越小对应于5d能级上更大的电子密度,即较低的氧化状态。图3a为参比样品金属Pt、H2PtCl6/EG溶液和PtO2粉末的XANES谱,从图中可以看到,H2PtCl6/EG溶液和PtO2粉末的白线峰强度远大于金属Pt的白线峰强度,这是由于H2PtCl6/EG溶液和PtO2粉末样品中的Pt物种处于Pt(IV)高价态。与H2PtCl6/EG溶液的白线峰强度相比,PtO2粉末样品的白线峰强度更高,且两者在11580 eV处具有不同的谱线形状,这是由O和Cl的配体效应不同所导致的。由于O的电子亲和性比Cl的电子亲和性强,Pt―O键的形成使Pt原子5d轨道上电子态密度降低更多,因此白线峰的吸收强度更高。
图3b-c显示了Pt胶体纳米簇形成过程中PtL3边XANES随反应温度及时间的变化,其中图3c是11565 eV附近的吸收峰。从图3c中可以观察到,室温下H2PtCl6和NaOH的EG溶液的白线峰强度低于H2PtCl6的EG溶液,说明混合溶液中Pt的平均价态低于Pt(IV),由此可以推测出室温下部分Pt(IV)已经被还原至Pt(II)。
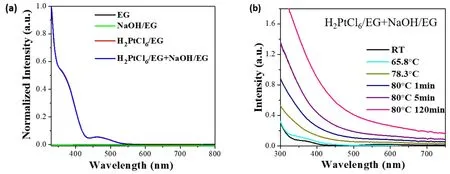
图2 UV-Vis吸收光谱测试结果。(a) H2PtCl6乙二醇溶液、NaOH乙二醇溶液及其混合溶液的UV-Vis吸收光谱;(b) Pt胶体纳米簇形成过程中的原位UV-Vis吸收光谱Fig.2 UV-Vis absorption spectra.(a) The absorption spectra of H2PtCl6 and NaOH in ethylene glycol (EG), and the mix solution of H2PtCl6/EG and NaOH/EG.(b) The in situ absorption spectra of reaction mixture for preparing unprotected Pt nanoclusters during reduction.
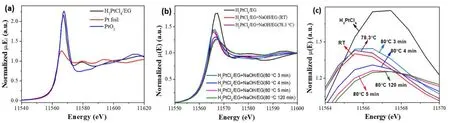
图3 Pt L3边XANES谱。(a)参比样品金属Pt、H2PtCl6/EG溶液和PtO2粉末;(b)非保护型Pt胶体纳米簇形成过程中XANES的变化图;(c) 11565 eV处吸收峰的放大图Fig.3 Pt L3-edge XANES spectra.(a) The reference of Pt foil, H2PtCl6 glycol solution, and PtO2 powder; (b) Time-evolution of reaction mixture for preparing Pt nanoclusters; (c) The enlarged image of the absorption peak at ca.11565 eV.
当温度升至78.3 °C时,反应体系的白线峰强度明显增强,说明Pt周围的配位环境发生了变化,对应于OH-配体取代Cl-配体的过程。由反应体系在78.3 °C下的UV-Vis吸收光谱可知,该温度下中间产物已缩合形成纳米粒子。在80 °C下继续加热反应体系,白线峰强度快速降低至一个基本不变值,暗示上述纳米粒子已被还原,生成了非保护型Pt金属纳米簇。
图4为参比样品和反应体系的 EXAFS傅立叶变换径向分布函数图。由图4a可知,位于1.64、1.96和2.47 Å (1 Å = 0.1 nm)处的峰(未校正相移)分别对应Pt―O键、Pt―Cl键和Pt―Pt键(相移校正后的键长分别为2.00、2.32、2.77 Å)。图4b为不同条件下反应体系的径向分布函数,对径向分布函数进行拟合得到Pt的不同配体的配位数列于表1。
从表中可以看到,初始反应体系中Pt周围Cl-的配位数为5左右,说明反应体系中部分已被还原为导致Pt离子周围Cl-的平均配位数小于H2PtCl6/EG溶液中Pt周围Cl-的配位数6。在反应温度由室温升至80 °C的过程中,Pt―Cl键的比例逐渐减少,Pt―O键形成且比例逐渐增加,说明体系中Pt周围Cl-逐渐被OH-所取代。随着反应在80 °C下进行,体系中开始有Pt―Pt键形成,Pt―O和Pt―Cl键的信号逐渐消失,对应于氧化物纳米粒子被还原为非保护型Pt金属纳米簇的过程。
以上分析结果表明,H2PtCl6前驱体在NaOH/EG溶液中易发生还原反应,常温下搅拌反应混合液,部分Pt(IV)就被能还原至Pt(II),使反应体系中同时存在Pt(IV)和Pt(II)。在加热升温过程中,氯化物前驱体与OH-反应,Pt周围配位的Cl-离子被OH-取代,且在Pt―Pt键形成前,反应体系UVVis吸收光谱中出现明显的纳米粒子的散射信号,说明中间产物已缩合形成了含Pt―O―Pt键的纳米粒子。随着反应的继续进行,Pt氧化物纳米粒子被还原,形成非保护型Pt金属纳米簇。
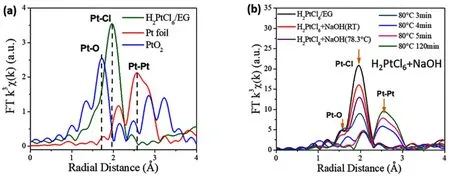
图4 k3权重的Pt L3边EXAFS傅里叶变换(径向分布函数)。(a)金属Pt、H2PtCl6/EG溶液和PtO2粉末;(b)非保护型Pt胶体纳米簇形成过程中径向分布函数变化图Fig.4 Fourier transforms of k3-weighted Pt L3-edge EXAFS spectra (Radial distribution function).(a) The reference of Pt foil, glycol solution of H2PtCl6, and PtO2; (b) The reaction mixture for preparing unprotected Pt nanoclusters under different conditions.
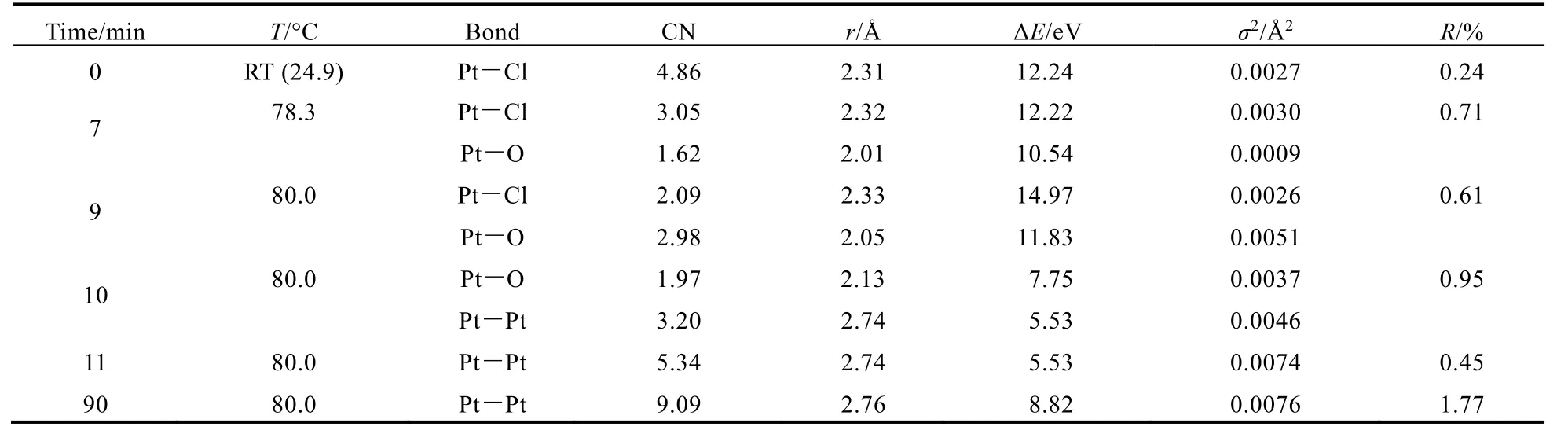
表1 由Pt L3 边 EXAFS光谱拟合得到的非保护型Pt纳米簇制备过程中Pt―Cl、Pt―O和Pt―Pt键的拟合参数Table 1 Fitting parameters of the Pt―Cl, Pt―O, and Pt―Pt bonds obtained from the Pt L3-edge EXAFS spectra of reaction mixture for preparing unprotected Pt nanoclusters.
图5为Pt胶体纳米簇形成过程中间物种的TEM照片和粒径分布图。从图中可以看到,反应初期获得的纳米粒子尺寸较大,且在透射电镜下衬度较低。其中,80 °C下加热反应物15 min获得的纳米粒子的平均粒径为3.7 nm (图5a),高分辨透射电镜(HRTEM)图显示其晶面间距为0.249 nm,远大于Pt(111)晶面间距(0.227 nm),说明此时反应体系中的纳米粒子是中间产物氧化铂。
随着反应进一步进行,纳米粒子的平均尺寸减小至2.4 nm (图5b),且图中存在衬度深浅不一样的两种纳米粒子,对应中间氧化物纳米粒子和Pt金属纳米簇。当反应时间进一步延长,体系中纳米粒子的平均尺寸减小至1.4 nm (图5d),此时纳米粒子的晶格间距与Pt(111)晶面的0.227 nm值相同,表明最终产物为Pt金属纳米簇。当氧化物被还原为金属纳米簇时,纳米粒子的密度增加而体积会减小。同时在TEM照片中,也观察到一个氧化物纳米粒子中可以形成多个金属纳米簇的情况。因此,相比初始纳米粒子的尺寸,最终得到的金属纳米簇的平均尺寸明显减小。
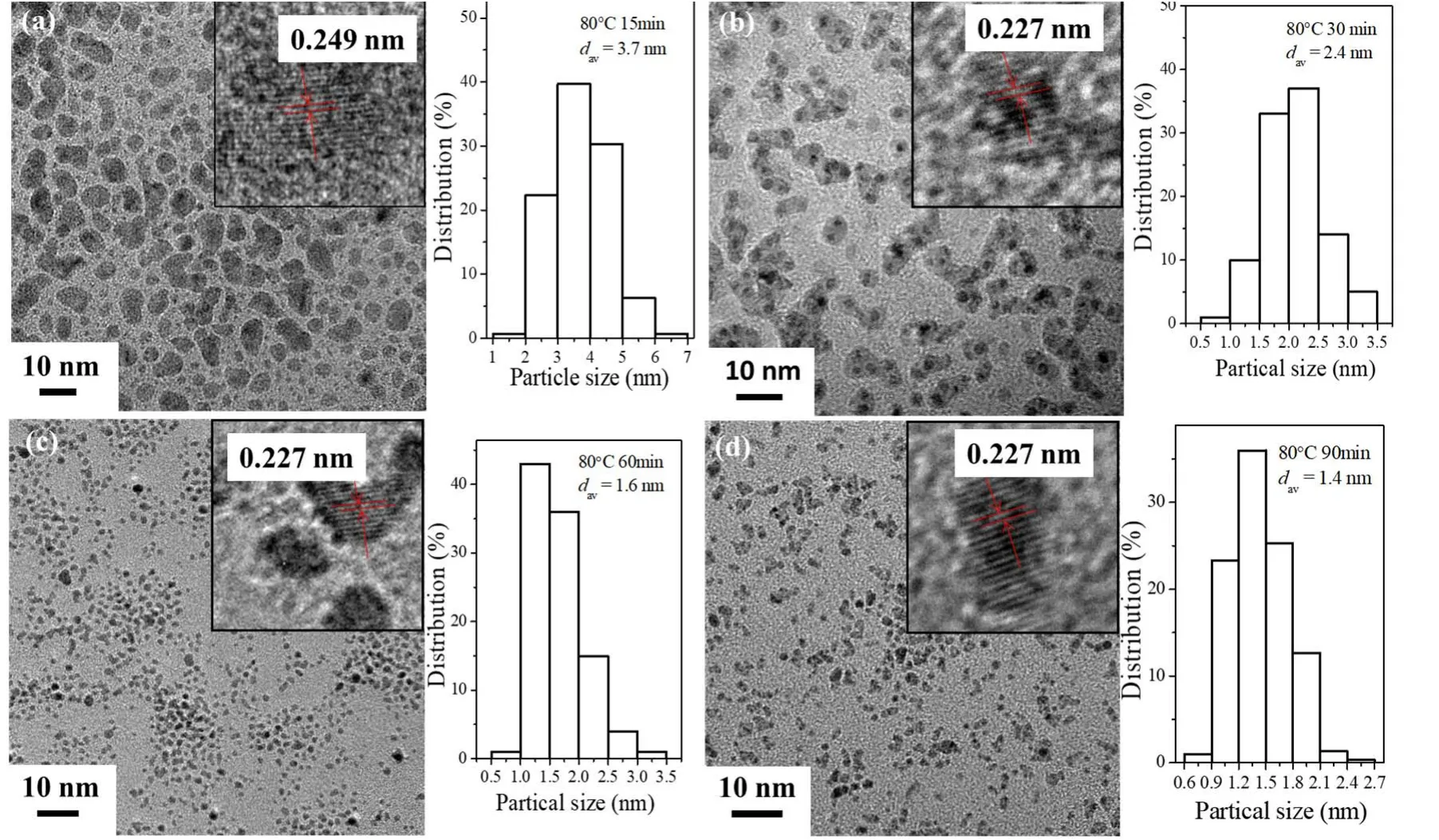
图5 80 °C下反应(a) 15 min;(b) 30 min;(c) 60 min;(d) 90 min过程中胶体纳米簇的TEM图像和粒径分布。插图为高分辨电镜图Fig.5 TEM images and particle size distributions of colloidal nanoclusters formed under 80 °C for(a) 15 min; (b) 30 min; (c) 60 min; and (d) 90 min.Inserts are HRTEM images.
图6为反应中间物种的选区电子衍射图(SAED),从图6a中可以看到,中间物种(80 °C下反应15 min)的SAED图中有清晰的衍射环,其对应的晶面间距分别为:0.250、0.215、0.154、0.130 nm,这些晶面间距与Pt纳米簇的晶面间距[0.227 nm(111)、0.140 nm (200)]不相符。80 °C下反应90 min后,所形成纳米粒子的SAED图(图6b)对应的衍射晶面间距与Pt纳米簇的晶面间距相符,以上实验结果与前述原位实验结果一致。
我们利用动态光散射(DLS)分析了反应体系中纳米粒子的尺寸变化,结果列于表2。从表中可以看到,由DLS测得的反应混合溶液中纳米粒子的数均粒径随反应时间的增加而减小,平均尺寸的变化趋势与TEM观察结果一致。此外,DLS测得的纳米粒子平均尺寸较大,这是由于所使用的动态光散射仪对大尺寸纳米粒子较为敏感。此处TEM和DLS测试结果统计的是体系中所有纳米粒子的平均尺寸,与最终生成的Pt金属纳米簇的平均粒径有所区别。
3.2 非保护型Ru胶体纳米簇的形成机理

图6 80 °C下反应(a) 15 min、(b) 90 min过程中胶体纳米簇的的SAED图Fig.6 The SAED pattern of nanoparticles in the reaction mixture for preparing Pt NCs under 80 °C for (a) 15 min; (b) 90 min.
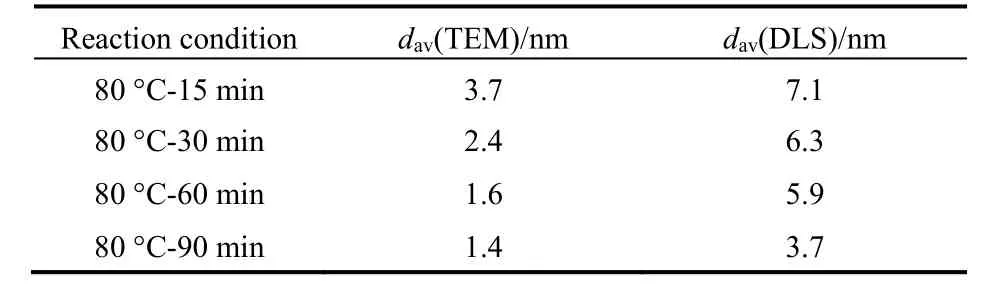
表2 由TEM和DLS测得的Pt纳米簇制备过程中纳米粒子的平均粒径Table 2 The average particle sizes of colloidal nanoparticles in the reaction mixture for preparing Pt nanoclusters measured by TEM and DLS.
图7a为RuCl3/EG溶液和RuCl3-NaOH/EG溶液的UV-Vis吸收光谱。从图中可以观察到,RuCl3/EG溶液在392 nm处有明显的吸收峰,对应于钌离子配合物的d-d电子跃迁。当RuCl3/EG与NaOH/EG混合时,混合溶液的吸收峰蓝移至346 nm,说明溶液中钌离子周围配位环境发生了变化,可能是RuCl3与OH-反应形成了钌的羟基配合物。图7b为非保护型Ru纳米簇制备过程中的原位UV-Vis吸收光谱。从图中可以看到,随着反应的进行,346 nm处的吸收峰强度逐渐降低,对应于体系中Ru的羟基配合物浓度逐渐降低。
当反应体系在100 °C下保持24 min时,可以观察到吸收光谱中有明显的纳米粒子的散射信号,说明此时反应体系中存在能被UV-Vis吸收光谱检测到的纳米粒子。
图7c为参比样品及RuCl3-NaOH/EG溶液的RuK边XANES。从图中可以看到,RuCl3-NaOH/EG溶液的XANES吸收峰不同于RuCl3/EG溶液,而与RuO2的峰相似,表明RuCl3与OH-发生了化学反应,钌离子周围由OH-配位,这与UV-Vis吸收光谱中出现的吸收峰蓝移的现象一致。图7d为非保护型Ru胶体纳米簇形成过程中反应混合物的原位XANES。从图中可以看到,反应初始阶段(体系在100 °C下反应前24 min),XANES的吸收信号变化很小,说明体系中Ru(III)的价态没有明显变化。继续在100 °C下加热6 min (100 °C下反应30 min),XANES吸收边后第一个峰的吸收强度明显减弱,说明钌离子的价态有所降低,即发生了还原反应。
图8为参比样品和反应体系的EXAFS傅里叶变换径向分布函数,拟合得到反应过程中各化学键的参数变化,列于表3。从表中可以看到,初始反应体系中主要以Ru―O键为主,说明RuCl3/EG和NaOH/EG混合溶液中RuCl3与OH-发生了反应,Ru离子周围以OH-配位为主,形成羟基配合物结合UV-Vis吸收光谱可知,在Ru―Ru键形成之前,散射信号已出现,说明进一步缩合形成氧化物纳米粒子。随着反应的进行,Ru―O的配位数逐渐减小且键长逐渐增大,说明随着还原过程的进行,Ru离子的价态逐渐降低。
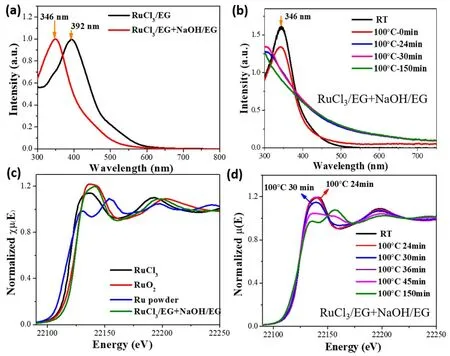
图7 反应过程中胶体纳米簇的原位表征。(a)反应前驱体的UV-Vis吸收光谱;(b) Ru胶体纳米簇形成过程中的UV-Vis吸收光谱;(c)参比样品Ru粉末片,RuCl3/EG溶液和RuO2及RuCl3-NaOH/EG溶液的Ru K边XANES光谱;(d) Ru胶体纳米簇合成过程中反应体系的Ru K边XANES光谱Fig.7 In situ characterization of colloidal nanoclusters.(a) UV-Vis absorption spectra of RuCl3 glycol solution and RuCl3-NaOH/EG solution; (b) The in situ UV-Vis absorption spectra of reaction mixture for preparing unprotected Ru nanoclusters under different condition; (c) Ru K-edge XANES spectra of Ru powder, RuCl3 glycol solution, and RuO2 powder; (d) Time evolution of Ru K-edge XANES spectra of reaction mixture for preparing unprotected Ru nanocluster under different conditions.
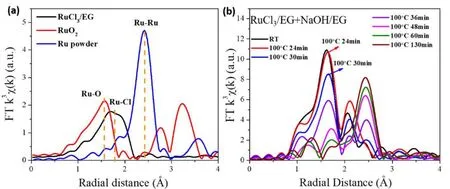
图8 k3权重的Ru K边EXAFS傅里叶变换(径向分布函数)。(a)参比样品Ru,RuCl3/EG溶液和RuO2;(b)非保护型Ru胶体纳米簇形成过程中径向分布函数的变化图Fig.8 Fourier transforms of k3-weighted Ru K-edge EXAFS spectra (Radial distribution function).(a) The reference of Ru powder, RuCl3 glycol solution, and RuO2 powder; (b) The reaction mixture for preparing unprotected Ru nanoclusters under different conditions.
图9为Ru胶体纳米簇形成过程中间物种的TEM照片、粒径分布图和SAED图。从图中可以看到,Ru胶体纳米簇形成过程中胶体纳米簇的平均粒径先逐渐增大随后逐渐减小。其中,反应体系在100 °C下反应5 min获得的纳米粒子的平均粒径为5.4 nm,且SAED衍射晶面间距对应Ru氧化物纳米粒子。反应体系在100 °C下反应120 min后,反应产物的TEM中观察到纳米粒子的平均粒径为1.4 nm,此时SAED衍射晶面间距对应Ru金属纳米簇,说明反应体系已完全还原。HRTEM照片显示,中间物种的晶面间距为0.253 nm (图9f),大于Ru金属纳米簇的晶面间距0.214 nm (图9g)。
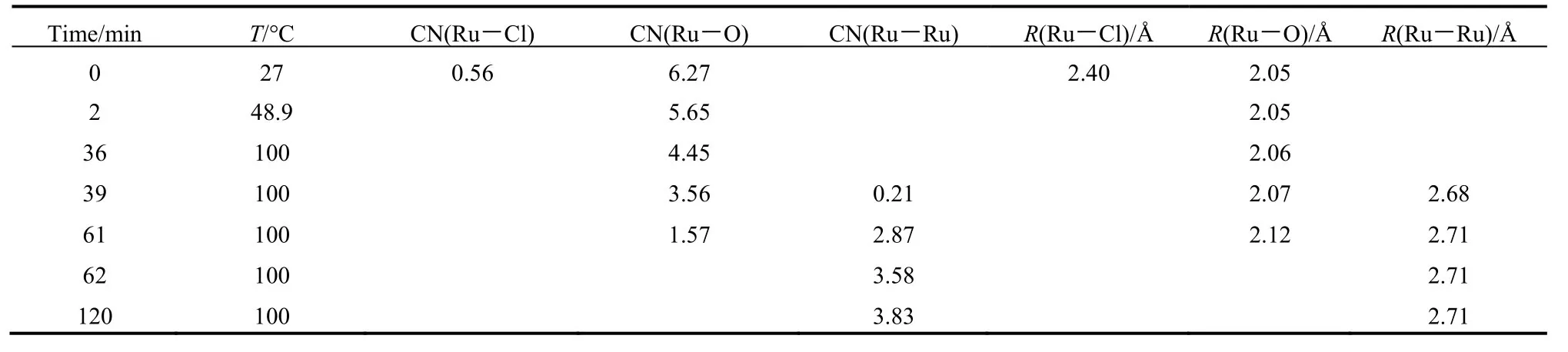
表3 由Ru K边EXAFS光谱拟合得到的非保护型Ru纳米簇形成过程中Ru―Cl、Ru―O和Ru―Ru键的拟合参数Table 3 Fitting parameters of the Ru―Cl, Ru―O, and Ru―Ru bonds obtained from the Ru K Edge EXAFS spectra of the reaction mixture for preparing unprotected Ru nanoclusters.
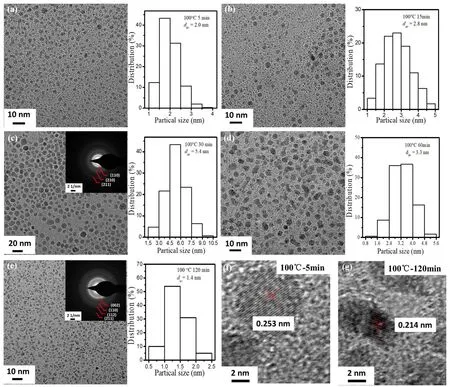
图9 100 °C下反应过程中胶体纳米簇的TEM图像和粒径分布:(a) 5 min;(b) 15min;(c) 30 min;(d) 60 min;(e) 120 min。100 °C下反应(f) 5 min及(g) 120 min后纳米粒子的HRTEM图。插图为SAED图像Fig.9 TEM images and particle size distributions of colloidal nanoclusters formed under 100 °C: (a) 5 min; (b) 15 min;(c) 30 min, (d) 60 min, (e) 120 min.HRTEM images of colloidal nanoclusters formed under 100 °C for (f) 5 min and(g) 120 min.Inserts are SAED patterns.
4 结论
本文采用原位快速扫描X射线吸收精细结构谱(QXAFS)和原位UV-Vis吸收光谱研究了非保护型金属胶体纳米簇的形成过程。结果表明在NaOH的乙二醇溶液中,H2PtCl6前驱体易还原,室温下即有部分Pt(IV)还原至Pt(II)。随着反应温度的升高,OH-逐渐取代了与Pt离子配位的Cl-,相应的原位XAENS中白线峰的吸收强度有所增加。综合原位外-可见吸收光谱、原位EXAFS、TEM以及DLS的表征结果可知,在Pt-Pt键形成之前,反应体系的UV-Vis吸收光谱中可观察到明显的纳米粒子的散射信号,原位EXAFS分析表明Pt纳米簇是由Pt氧化物纳米粒子还原所形成的;在碱-乙二醇法合成非保护型Ru金属纳米簇的过程中,OH-首先取代了RuCl3中的Cl-,形成羟基配合物后者进一步缩合形成氧化钌纳米粒子。Ru金属纳米簇是由乙二醇还原氧化钌纳米粒子形成的。由于先形成了氧化物纳米粒子,后续的还原反应被限制在氧化物纳米粒子内,使最终得到的非保护型金属纳米簇具有尺寸小、分布窄的特点。
Acknowledgements:The authors thank the Photon Factory Advisory Committee (KEK, Japan) for approval of EXAFS measurements (Proposal No.2017G029).
Supporting Information:available free of chargeviathe internet at http://www.whxb.pku.edu.cn.
猜你喜欢
杂志排行

物理化学学报的其它文章
- Vapor-Liquid-Solid Growth of Bi2O2Se Nanoribbons for High-Performance Transistors
- 四卤化锰(II)配合物的结构调控及力致发光性能
- Conformational Switching of Verdazyl Radicals on Au(111)
- Photocatalytic CO2 Reduction Using Ni2P Nanosheets
- Swelling Characteristics of g-C3N4 as Base Catalyst in Liquid-Phase Reaction
- 聚合物支撑的金属有机骨架膜的制备及其气体分离性能