X射线晶体学结合电子晶体学在复杂无机晶体结构解析中的应用
2020-04-02黎建林聪林建华孙俊良
黎建,林聪,林建华,孙俊良
北京大学化学与分子工程学院,北京 100871

1 引言
众所周知,物质的结构决定物质的物理化学性质,物理化学性质是物质结构的反映。只有充分了解物质的结构,才能深入认识和理解物质的性能,才能更好地改进化合物和材料的性质与功能,设计出性能优良的新化合物和新材料。因此,材料的原子结构对其性能的影响非常大。晶体学就是一门从实验上确定原子结构的科学。
需要观察到材料的原子结构,首先需要一个好的显微镜,显微镜光源的波长决定了其观察物质的分辨率。根据Rayleigh公式,

其中,r为显微镜的分辨率,λ为光源的波长,μ和α为显微镜常数。因此对于光学显微镜来说,其波长为400-700 nm,其最大分辨率在200 nm。材料中原子的距离大概为0.1-0.2 nm,远远超出了光学显微镜所能观察的精度范围。可用于晶体学分析材料原子级结构的光源主要有三种:X射线(铜靶波长为1.5406 Å (1 Å = 0.1 nm)),电子(加速电压为200 kV的波长为0.0251 Å)和中子(热中子波长约为0.5 Å)。中子由于存在偶极矩,因此也可以用于分析材料的磁结构。本文主要讨论X射线和电子作为光源在晶体学结构解析中的应用。
晶体学解析结构的原理可以用以下两个公式进行描述:
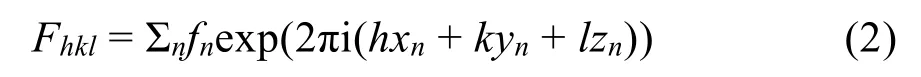
其中fn为第n个原子的散射因子;(xn,yn,zn)为原子位置,hkl为衍射指标。由公式(2)可以计算得到晶体结构的结构因子,结构因子由振幅和相位组成。

其中x,y,z为原子在晶胞中的分数坐标。Vcell为晶胞体积,|Fhkl|为衍射指标hkl的结构因子振幅,φhkl为衍射指标hkl的结构因子相位。由公式(3)可以通过结构因子计算得到电子密度图(X射线)或者电子势能图(电子),在电子密度图(电子势能图)中,电子密度(电子势能)大的地方对应原子位置。
1.1 散射因子
X射线主要对核外电子进行散射,原子对X射线的散射强度就大约正比于原子序数。原子序数越大,核外电子越多,散射能力越强,因此X射线对重原子比较敏感,而对于核外电子非常少的氢,其位置在X射线中难以精确确定。由于核外电子云密度的分布随着半径增加而减少,将原子中不同空间位置对X射线散射的贡献叠加就是原子的散射因子f。除此之外,原子散射因子f的数值也与衍射角有关。轻重原子的散射因子f数值随着衍射角θ的增加而显著减少。但是轻原子的散射因子本身比较小,随着衍射角θ的增加,散射因子的数值变得更加小。而重原子对高θ角的衍射贡献相对较明显1。
电子由于带有电荷,不仅对核外电子进行散射,还对原子核进行散射。电子与原子核和核外电子主要以库伦相互作用。然而原子核非常小,且带正点荷,与带负点的电子云相比,原子核相当于一个点。因此电子与物质的相互作用非常复杂,既有弹性散射,也有非弹性散射。弹性散射在于与核外电子和原子核的相互作用中能量不发生变化,而非弹性散射在相互作用过程中,电子发生能量损失,只有弹性散射部分才用于结构分析。电子散射因子也随着散射角的增加而显著下降2。
1.2 X射线晶体学
X射线衍射晶体学是目前最重要的结构解析方法。X射线晶体的发展最早可以追溯到1912年,MAX Von Laue和其助手发现X射线衍射。1912-1913年Bragg父子提出Bragg方程。鉴于他们在晶体学中的贡献,MAX Von Laue和Bragg父子分别获得了1914年和1915年的诺贝尔物理学奖。在过去的一百多年中,非常多的材料的有序原子结构通过X射线衍射被解析出来,比如合金,陶瓷,半导体,甚至冰。通过对这些材料结构的深入分析,使研究其性质,推广该材料的应用成为可能。X射线衍射晶体学分为粉末X射线衍射(Powder X-ray diffraction,PXRD)和单晶X射线衍射(Single crystal X-ray diffraction,SCXRD)。
在X射线晶体学中,所有衍射峰的强度可以通过实验直接得到,衍射峰强度的平方根为结构因子振幅。但是通过衍射得到的数据中结构因子的相位是缺失的,通过公式(3)不能直接用来计算电子密度图。但是可以通过各种不同的算法提取其相位,比如帕特森法3,直接法4,电荷翻转法5,最大熵法6,基因算法7,模拟退火法8等等。单晶X射线衍射由于所有的衍射峰是独立的,且强度准确,相位可以通过各种算法准确提取出来,因此被认为是最快捷,最有效的结构解析方法。最近几年,很多新分子筛结构通过单晶X射线衍射直接解析出来,比如,SU-15(SOF)9,SU-32(STW)9,PKU-9(PUN)10等等。但是单晶X射线衍射对晶体质量要求非常高,需要的晶体大小在几个微米以上,因此对于很多难以长成大单晶的样品来说,单晶X射线衍射解析结构受到了很大限制。在某些情况下,即使晶体大小足够进行单晶X射线测试,但是由于材料中的无序或者缺陷仍然会使单晶X射线衍射解析结构困难重重。
在不能得到大单晶的情况下,粉末X射线衍射解析结构成为一种标准的结构解析方法。最近几年发现的新型硅基分子筛、锗酸盐,硼铝酸盐的结构大部分是通过粉末X射线衍射解析的,比如IM-5(IMF)11,IM-16(UOS)12,SU-7413,PKU-114,PKU-215。在粉末X射线衍射中,几百万个小晶体随机取向,在单晶X射线衍射中是一个衍射点的峰在粉末X射线衍射中形成了衍射环。每个衍射环的大小(衍射角,2θ)通过d值计算。通过扫描2θ,三维空间的倒易点阵被投影在了一维空间,因此若d值相近或者相等的衍射峰就会发生重叠。当粉末X射线衍射峰的重叠不是很严重的时候,可以在电荷翻转算法中通过FIPS算法等对粉末衍射峰的强度重新分配16,17。当晶胞参数大,在半峰宽为0.3范围内的峰重叠度超过80%时,很难通过纯数学算法获得重叠峰的强度,造成提取结构因子相位失败而导致结构解析失败。另外一方面,粉末X射线衍射是样品中所有物相的信号,因此若样品不纯也很难对未知相的结构进行解析。
1.3 电子晶体学
除了X射线晶体学外,电子晶体学也可以用来进行材料的结构解析。特别是对于那些用单晶X射线衍射来说晶体太小,粉末X射线太复杂材料,电子晶体学成为一种结构分析的重要手段。与X射线晶体学相比,电子晶体学的最大不同在于:(1)电子与物质的相互作用是X射线的几百万倍,使得电子可以用于研究比X射线衍射所需尺寸小得多的微米,纳米级的晶体;(2)电子束可以通过电磁透镜控制光斑的大小,高分辨像中得到确定未知晶体结构时所需要的结构因子相位。由于这些不同,使得电子晶体学在结构解析方面有着特殊的应用。
电子与物质的强相互作用在结构解析中既有利的一方面,也有弊的一方面。首先,单晶X射线衍射需要的晶体大小在几个微米左右,粉末X射线衍射对晶体大小的要求可以降低到50 nm,当晶体尺寸再小时,粉末衍射峰将发生宽化,导致更严重的峰重叠。而电子作为探针(光源)时,几个晶胞参数大小的晶体产生的信号足够用来进行结构解析。类似单晶的电子衍射数据能直接在多晶样品中获得。传统收集电子衍射数据的方法是手动倾转晶体,收集不同晶带轴方向的电子衍射数据,这种方法费时费力,且要求实验者具有很高的电子显微镜操作水平。近几年来发展的三维电子衍射技术,如自动电子衍射断层扫描(automated electron diffraction tomography,ADT)18,旋转电子衍射(rotation electron diffraction,RED),连续旋转电子衍射(continuous rotation electron diffraction,cRED)19,电子衍射收集实现了自动化,且收集时间缩短到了几分钟。利用这种三维电子衍射技术,大量的、各种类型的材料的结构被解析出来。电子与物质的强相互作用对结构解析不利的一方面在于动力学效应和电子辐照损伤。由于强相互作用,电子在通过材料时不止发生一次衍射。电子衍射不再是运动学的,而是动力学,从而导致电子衍射强度与结构因子的强度不再是简单线性关系,这样使得通过电子衍射解析结构比单晶X射线衍射更加困难。虽然新发展的3D电子衍射技术(ADT,RED)能大大降低动力学效应,很多复杂结构仍很难获得初始结构模型。当电子与材料相互作用时,电子将把能量传递给材料,从而破坏材料中的化学键,并缓慢加热样品,导致材料结晶性下降,这种称之为辐照损伤。因此对于电子束敏感样品,如分子筛20,21,金属簇框架材料22,化学键有机框架材料23-25等,在数据收集过程中需要特殊保护样品,最大限度的降低电子对材料的破坏。
除了衍射模式,透射电子显微镜还可以进行成像模式,由于电子的波长非常短,因此在成像模式下得到的照片分辨率可以在1 Å以上,可以直接观察到晶体结构中某个方向的原子柱,因此被称为高分辨透射电子显微镜(High resolution transmission electron microscopy,HRTEM)照片。所有的衍射技术仅仅能得到结构因子中的振幅信息,而在HRTEM照片中可以通过傅里叶转换直接得到结构因子的相位和振幅的信息。单张HRTEM照片只能得到2个方向倒易空间的振幅和相位信息,因此需要结合不同正带轴的HRTEM照片重构出三维电子势能图,从而确定原子结构。利用这种方法很多复杂的分子筛结构被解析出来11,26-30。HRTEM照片最大的问题是由于电子辐照损伤,对于很多材料来说,很难获得原子分辨率的照片。
综上所述,电子晶体学和X射线晶体学均是非常强大的结构解析手段,但是均存在各自的不足(表1为单晶X射线衍射,粉末X射线衍射,HRTEM照片和电子衍射的比较)。而对于复杂结构,通过单一的一种技术,往往很难达到结构解析的目的,需要多种技术相结合的方法才能最终完成结构的确定。在下面的章节中,我们将主要讨论X射线晶体学结合电子晶体学在粉末X射线衍射峰重叠,样品不纯,骨架结构存在无序,客体分子存在无序,赝对称性,非共度结构等复杂结构问题中的应用。
2 粉末X射线衍射峰重叠
峰重叠是粉末X射线衍射结构解析的一个主要问题。峰的重叠使得衍射强度提取不准,从而造成结构解析失败。虽然发展了很多重新分配重叠峰的方法,但是对于特别严重的峰重叠问题(半峰宽为0.3时的重叠度超过80%),即使对于高分辨率的同步辐射数据,简单的分配方法不是特别有效。因此需要其他技术辅助进行结构解析。
2.1 HRTEM照片与粉末X射线衍射相结合
正如上文所述,X射线衍射的方法只能得到结构因子中的振幅,所有的相位是丢失的。而相位可通过细致的图像衬度传递函数(contrast transfer function,CTF)提取获得,将提取的相位与粉末X射线衍射相结合能够大大提高结构解析的成功率。目前这种将正空间和到空间信息结合起来用于结构解析的算法有Focus31和电荷翻转法(Charging Flipping)。Focus是专门用于分子筛结构解析的算法。高硅的IM-511和TNU-926分子筛就是通过将HRTEM照片得到的相位信息分别代入Charging Flipping和Focus中结合粉末X射线衍射解析结构。TNU-9是非常复杂的分子筛之一,晶胞参数大,对称性低,且晶体尺寸小,PXRD峰重叠特别严重,同步辐射PXRD在0.3的半峰宽下峰重叠度超过85%。因此通过PXRD解析其结构非常困难。用CRISP软件提取了三个方向的HRTEM照片中的258个衍射峰的相位信息,结合PXRD数据代入Focus中,经过16天的时间计算成功的解析了其结构,该结构有24个独立的硅原子,具有3维10圆环孔道结构。

表1 单晶X射线衍射,粉末X射线衍射,HRTEM照片和电子衍射的比较Table 1 Comparison of SCXRD, PXRD, HRTEM and ED.
IM-5是另外一个非常复杂的高硅分子筛,正交对称性,85%左右的峰重叠。收集3个主带轴的HRTEM照片,得到了95个衍射峰的相位,将HRTEM照片得到的相位信息作为初始相位代入电荷翻转算法时,发现得到的电子密度图很难用于硅原子位置的确定。随后,通过HRTEM照片重构得到一个空间群为C2cm的粗略模型,将该模型计算得到的相位作为初始相位再次代入电荷翻转算法中,其中25%的相位为随机相位,成功的得到了五个可能的结构模型。最后通过Rietveld精修发现IM-5具有更高的Cmcm对称性的二维孔道分子筛。
除此之外,HRTEM照片也可以辅助用于模型构建与低分辨率PXRD相结合解析结构。铝硼酸盐PKU-215的PXRD由于分辨率低,晶胞参数大(六方:a= 30.489(2) Å,c= 7.013(1) Å),很难通过PXRD解析其结构。但是,PKU-2的c轴的晶胞参数和c方向的HRTEM投影与另外一个铝硼酸盐PKU-1高度相似(R-3,a= 22.038 Å和c= 7.026 Å)。PKU-1是一个由AlO6八面体构成的18圆环直形孔道结构,因此,根据PKU-114的模型和PKU-2的HRTEM照片,PKU-2的结构模型很快被构建出来,并通过Rietveld进行确认(图1)。
2.2 PXRD与电子衍射相结合
我们知道收集HRTEM照片对电镜操作者的要求非常高,特别是对于电子束敏感样品,且费时费力。而电子衍射相对来说容易一些,且电子剂量低,因此电子衍射与PXRD结合也是一个非常实用的解析复杂化合物结构的方法。
电子衍射与PXRD相结合解析复杂结构最直接的方法是根据电子衍射的强度对PXRD的重叠峰进行分配。ITQ-3732是一个非常复杂的硅锗酸盐,立方晶系(P4132,a= 26.5126 Å),在晶面间距d> 1.2 Å下的峰重叠度高达94%。且由于含锗,电子束非常敏感,高分辨电镜照片的分辨率只有6 Å。因此收集了[100]、[110]、[111]和[120]四个方向选区电子衍射照片,通过选区电子衍射的强度对PXRD的重叠峰进行重新分配,改进后的PXRD代入charge flipping算法中显著提高了电荷翻转迭代法收敛的速度,得到的电子密度图如图2a所示。ITQ-37的最终Rietveld精修结果确定ITQ-37为具有30圆环的手性超大孔分子筛(图2b)。除此之外,三维电子衍射数据(3D ED)也可以用于PXRD重叠峰的分配。如微孔铝硼酸盐PKU-333,具有三方对称性,在1 Å分辨率下,半峰宽为0.2时的峰重叠度超过86%。孔道中无序的Cl离子和硼原子的配位环境多变,均增加了结构解析的难度。在这个工作中,根据3D ED数据重新分配PXRD的强度,采用直接法直接得到了大部分骨架原子和孔道中的氯原子(图2c)。
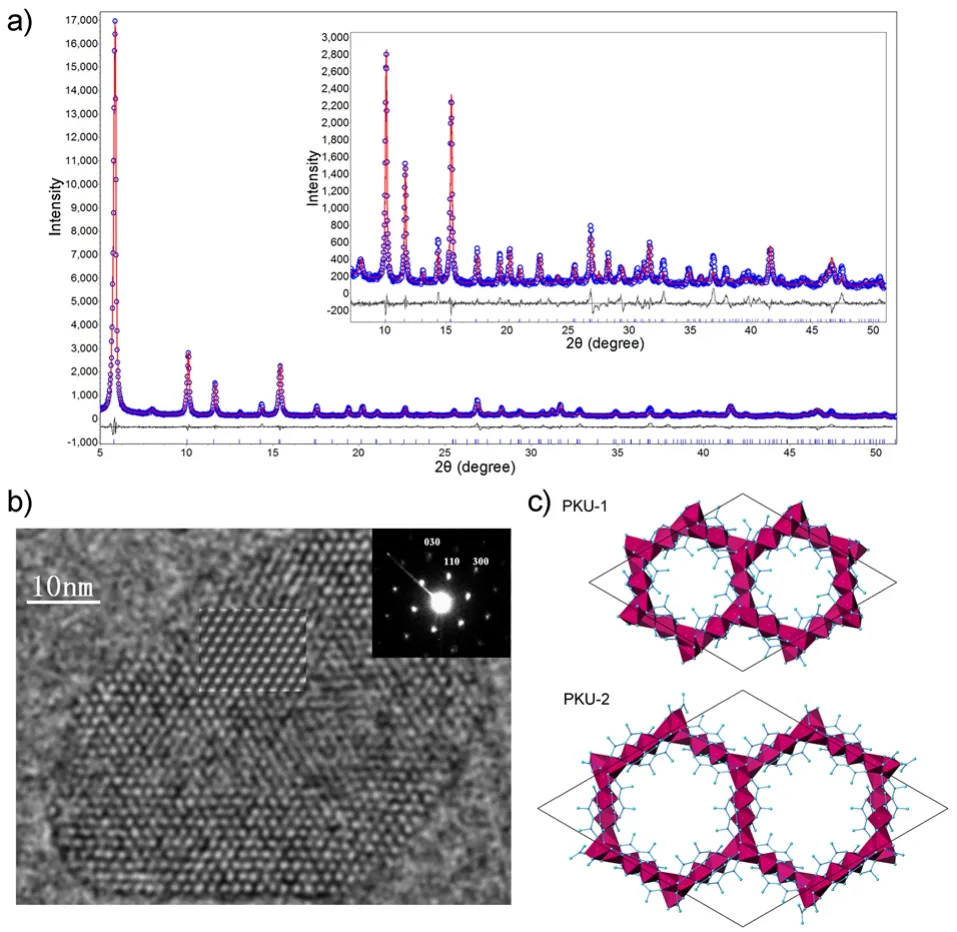
图1 具有大孔径的微孔材料磷铝酸盐的结构确定14,15Fig.1 The structure determination of microporous aluminoborates with large channels14,15.
3 样品不纯
对于合成化学家和矿物学家来说,发现新材料是一件非常兴奋的事情。但是合成受很多因素影响,比如原料、化学组分、反应温度、反应时间等,均需要严格优化,这个优化过程费时费力。在初始合成和发现过程中,往往很难获得纯相,如果能在不纯的样品中快速得到新相的结构,从而调整合成策略,能够大大缩短发现新材料的周期。三维电子衍射技术正好可应用于快速物相分析和结构。三维电子衍射可用来分析纳米级晶体的结构,它们可以利用与X射线衍射类似的算法得到原子位置(直接法4、Patternson法3、电荷翻转法5、最大熵法6,基因遗传法7等),且样品不纯也没有任何影响。首先通过三维电子衍射解析得到粗糙的结构模型,根据结构模型对合成条件进行修正,获得纯的物相。最后通过粉末 X 射线衍射、粉末中子衍射精修、固体核磁等其他技术相结合获得精确的结构模型。

图2 ITQ-37和PKU-3的结构模型32,33Fig.2 The structure model of ITQ-37和PKU-332,33.
BiTi0.855Fe1.145O4.93、BiTi4FeO11、BiTi2FeO734三个新化合物在发现过程中均非主相,在初始合成过程中随机选择7个原料组分比例进行高温固相合成,通过粉末X射线衍射分析发现T1、T2、T4、T6的衍射峰均可被已知化合物指标。而T3、T5、T7样品的粉末X射线衍射存在大量的不能用已知化合物的特征峰进行指标的衍射峰,考虑到T3和T5、T7组分的巨大差异,因此在这个体系中至少有两个结构未知的化合物。由于粉末X射线衍射很难对T3、T5、T7中的未知相的结构进行解析,因此利用RED技术收集了每个样品中10-15个500 nm左右单晶颗粒的三维电子衍射数据(图3)。对所有三维电子衍射数据进行三维重构发现,有三套不同的晶胞参数不能在无机晶体结构数据库中检索到。因此认为在这个体系中发现了三个新结构的新化合物。随后采用单晶X射线衍射结构解析的方法,利用三维电子衍射的数据对T3、T5、T7中的未知相的结构进行解析。根据结构模型和电荷平衡关系很快确定了三个新化合物的分子式为BiTi0.855Fe1.145O4.93、BiTi4FeO11、BiTi2FeO7,并修正合成时的投料比例得到了三个化合物的纯相。同样的,硅锗分子筛PKU-16在初始发现过程中也是非主相,且尺寸只有40-200 nm。通过RED技术收集的3D ED数据直接解出了其结构模型,随后调整合成条件得到了其纯相,PKU-1621是一个具有11 × 11 × 12圆环孔道的新型硅锗分子筛(图4)。因此3D ED方法是对不纯样品的强大的物相分析和结构确定的方法。
4 骨架结构存在无序

图3 通过快速物相分析与结构确定发现新型复杂金属氧化物34Fig.3 Discovery of novel complex metal oxide materials by rapid phase identification and structure determination34
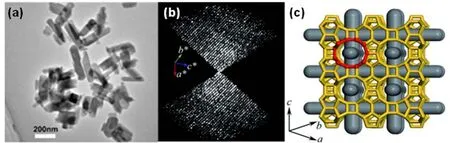
图4 通过电子晶体学解析具有11 × 11 × 12圆环孔道的硅锗分子筛PKU-1621Fig.4 PKU-16: A germanosilicate structure with 11 × 11 ×12-ring channels solved by electron crystallography21.
骨架结构的无序主要分为共生和堆积无序,在矿物、分子筛和无机开放骨架材料中骨架的无序非常普遍。由于不能用传统的结构确定方法解析其结构,骨架结构无序材料的结构确定目前来说还是一个非常大的挑战。对于骨架无序的分子筛常见的结构确定方式是通过已知结构猜测其无序结构模型,但很多情况下其结构确定需要PXRD、SCXRD、HRTEM等多种技术手段结合才能最终确定其真实结构(比如beta分子筛35-38、ITQ-3927、ABC家族分子筛39、ITQ-3340等)。因此骨架结构无序材料的结构确定是一个非常大的挑战。
Beta分子筛是目前广泛被工业应用的沸石分子筛,1967年美孚公司首次合成得到41,但是其结构直到1988年才正式确定36。到目前为止beta分子筛家族有五种晶型(polymorph),它们均是通过一个相同的层以不同的堆积方式构成。常见beta分子筛是由polymorph A和Polymorph B共生,其结构最初是通过HRTEM和ED相结合的方法确定。Beta分子筛的polymorph C是2000年首次被合成出来,其结构通过单晶X射线衍射确定37。Beta分子筛的polymorph D和E在2012年首次被合成出来35,虽然晶体大小在30 μm左右,但是由于孪晶和严重的无序问题,同步辐射SCXRD仍然不能确定其晶胞参数。为了确定其晶胞参数,在不同微小的单晶区域拍摄了系列选区电子衍射(SAED)得到了两套晶胞参数,分别为单斜晶系和正交晶系,随后根据选区电子衍射得到了晶胞参数对单晶X射线衍射进行指标化,发现单晶衍射存在4重孪晶。指标化后,只有h= 3n的衍射峰是锐锋,其他衍射峰由于骨架结构无序的存在导致拉长。因此,将0kl衍射峰提取出来构建a方向的结构,结合b方向的高分辨电镜照片最终确定了beta polymorph D和E的结构(图5)。
5 确定客体分子的位置
客体分子在开放骨架材料的合成与应用中有重要作用,比如分子筛,锗酸盐,磷酸盐,硼酸盐等。客体分子既可以作为开放骨架结构的结构导向剂,也可是稳定骨架结构的孔道填充剂。因此确定客体分子在开放骨架材料中的位置有助于研究材料的晶化机理,指导对骨架材料进行功能化,拓宽其应用。由于客体分子往往是小分子,散射弱,且客体分子的对称性往往与骨架结构的对称性不匹配,以无序的形式存在,即使是单晶X射线衍射也很难精确确定客体分子的位置。许多重要的开放骨架材料却只能得到多晶,很难获得高质量的单晶,使得确定客体分子位置更加困难。因此确定客体分子的位置需要多种技术相结合。
标准的确定客体分子的方式是在Rietveld精修过程中通过差值电子密度图来确定。有机结构导向剂对称性低,散射弱,而骨架结构对称性低散射强,这种巨大差异导致很难通过简单差值电子密度图来确定。模拟退火算法作为正空间的全局优化算法结合Rietveld精修可以确定有机结构导向剂的位置。具有开放骨架的锗酸盐SU-74-MPMD13和GeOJU9042是以2-methylpentamethylenediamine(MPMD)和1,5-bis(methylpyrrolidinium)作为结构导向剂合成得到。其骨架结构通过电荷翻转法确定,虽然骨架原子能被精修得非常合理,但是通过差值电子密度图很难确定客体分子的位置。正空间的模拟退火算法被证明是一个非常有效的确定客体分子位置的方法。将整个客体分子以刚性体的形式随机放入孔道中,随后基于模拟退火算法寻找其化学合理的位置,最后对骨架和客体分子进行Rietveld精修确定有机结构导向剂的最终位置(图6)。

图5 结合电子晶体学和单晶X射线衍射解析具有12圆环交叉孔道的新型共生的beta分子筛SU-7835Fig.5 Intergrown new zeolite beta polymorphs with interconnected 12-ring channels solved by combining electron crystallography and single-crystal X-ray diffraction35.
原位中子散射和粉末X射线衍射相结合也能确定客体分子的在开放骨架结构孔道中的位置。在NOTT-30043的结构中,通过原位的中子散射和粉末X射线衍射相结合,成功的确定了CO2和SO2在孔道中的位置,客体分子与骨架结构主要以弱氢键相互作用(图7)。除此之外,客体分子也可以用来确定骨架结构中的催化活性位点44,以有机客体分子为酸性位点的探针,将吡啶,甲醇等有机小分子吸附在H-ZSM-5分子筛的孔道中,通过原位的同步辐射PXRD精修得到孔道中小分子的位置,从而在原子水平上确定H-ZSM-5分子筛酸性位点的位置。

图6 通过粉末电荷翻转法和模拟退火法确定复杂的开放骨架结构的锗酸盐化合物13Fig.6 The structure of a complex open-framework germanate obtained by combining powder charge-flipping and simulated annealing13.
6 孪晶和赝对称结构
赝对称结构往往伴随着孪晶,导致很难判断正确的空间群。在孪晶中,一个晶体同时存在两个或者多个点阵结构相同的晶畴,这些晶畴取向不同或者互为镜像。最常见的孪晶是非缺面对称孪晶,孪生的两种晶格不互相重叠,因而倒易晶格和衍射点基本不重叠。另外一种非常复杂的孪晶叫缺面孪晶或者赝缺面孪晶,衍射点完全重叠或者近似完全重叠。因此,晶体中存在赝对称性是结构解析中的又一难题。

图7 通过原位的中子散射和粉末X射线衍射相结合确定了CO2和SO2在孔道中的位置43Fig.7 Locating carbon dioxide and sulfur dioxide in a decorated porous host by PXRD and inelastic neutron scattering43.
PKU-1445是一个具有三维大孔的新型硅锗分子筛,其结构由于存在严重孪晶和赝对称性,最终通过结合HRPXRD,RED,NMR和IR光谱成功解析其结构。虽然能获得20 μm大小的单晶,由于严重的孪晶,同步辐射单晶衍射没法得到正确的晶胞参数。因此为了避免孪晶,收集了3D ED数据。指标化重构的3D 倒易点阵得到的对称性为四方晶系(a= 19.2 Å,c= 29.6 Å,空间群I4/mcm)。然而,尝试对从PXRD和RED数据直接解析结构仍然没有成功。对比PKU-14和锗酸盐分子筛ASU-746和石英相GeO246的IR光谱发现,PKU-14可能是以GeO4单元链接的分子筛结构。以GeO4为结构单元通过平行退火算法构筑了由四个独立GeO4单元构成的可能结构模型,但是该模型的实验PXRD与模拟的PXRD在高角度区域差别非常大,且在结构精修过程中R因子非常高。考虑到F离子通常占据在双四元环结构基元中,PKU-14的19F MAS NMR光谱显示有四个信号,而以I4/mcm为空间群的四方结构模型只有两个对称性独立的F位置。因此,PKU-14的真实结构应该采用更低的空间群。在仔细分析同步辐射PXRD数据发现,实验室PXRD的第一个强峰(110)分裂成两个峰。因此,四方结构应该降到单斜结构,Y角偏离90°,最终结构模型修正为单斜结构a= 19.8075(7) Å,b= 26.7538 Å,c=19.8127 Å和β=90.4810(5)°,空间群I2/m。在单斜结构中有16个独立的锗原子,36个氧原子,且精修的Rwp因子显著提高,并收敛到11.30% (图8)。
7 非周期性结构
上述讨论的结构问题均是具有三维周期性的结构。还有一类非周期性结构,比如非公度调制结构,非公度共生化合物和准晶。调制结构是在普通周期结构上叠加一个微扰而形成的结构。叠加的微扰一般具有自己的固有周期,如果这个周期与晶体的原有周期成简单整数比,则形成公度的调制结构,即超结构;如果这个周期与晶体的原有周期不成简单整数比,即形成非公度调制结构;如果微扰本身无序,就形成无序结构或无定形结构47。非公度共生化合物与非公度调制结构不同,它没有基本的或平均的小晶胞,但可以把其整体结构拆分成几个具有各自周期的亚结构,任何两个亚结构的周期之间至少在某一方向上不成简单整数比,即至少有一维是非公度的。由于受到其他亚结构的作用(这种作用可以被看作具有不同周期的微扰),每个亚结构中的原子都会一定程度地偏离平均位置而形成调制结构.因而非公度共生化合物可以看成是多个非公度化合物共生在一个晶体内。准晶是另一类非公度结构,其特点是具有非传统晶体学的对称性,如5次、8次或10次对称性48,49。随着超空间群理论50和结构解析算法superflip的发展,许多非周期结构通过SCXRD被解析出来,如氧缺陷材料51,非线性光学材料52,合金材料53等。
三氯乙酸正异丙苯胺是一个新型有机铁电材料54。在150 K时为铁电相,单晶X射线衍射照片的主衍射点周围观察到一系列的卫星衍射点,且卫星衍射只分布在沿c轴方向。采用四个参数对衍射点进行指标H=ha*+kb*+lc*+mq,q= (0, 0,0.1589),由于q是不是简单有理数,因此该结构为非共度调制结构。对其衍射点进行指标发现其衍射规律为h0lm:h+m= 2n,0klm:l= 2n,hk00:k= 2n,确定其超空间群为Pbca(00g)0s0。利用superflip算法在超空间群下直接对其非公度结构进行解析(图9)。
图8 3D 12圆环孔道结构的分子筛PKU-14的结构模型45Fig.8 The model of PKU-14 with a 3D 12-ring zeolite45.

图9 有机分子铁电体中独特的长程有序非共度调制结构54Fig.9 Unusual long-range ordering incommensurate structural modulations in an organic molecular ferroelectric54.

图10 PbBiNb5O15的电子衍射图和结构模型55Fig.10 The electron diffraction pattern and structure model of PbBiNb5O1555.
而对于很难收集单晶衍射数据的非公度材料,PXRD和正带轴的ED的结合是一种标准的非公度材料结构分析的方法。PbBiNb5O15(PBN)55是一种四方钨青铜氧化材料,[11¯ 0]带轴方向的选区电子衍射的主衍射周围能观察到五阶卫星衍射,且调制矢量q为q=α(a* +b*) +γc* (α~ 0.3,γ~ 0.5),因此可能是(3 + 2)D四方钨青铜结构,晶胞参数为a=b~ 12.5 Å,c~ 3.9 Å (图10)。由于在α(a* -b*) +γc*方向没有观察到卫星衍射点,而在001带轴方向不符合(3 + 2) D四方对称性,因此PBN应该采用(3 + 1) D结构进行解析。根据二阶非线性信号和铁电性能,可能的空间群为极化空间群Cmm2,Cm2m,和C2mm。通过原位变温同步辐射PXRD确定极轴为b轴,超空间群为Cm2m(α, 0, 0.5)000 (a=17.65852(10) Å,b= 17.67709(10) Å,c=3.87645(3)Å),q矢量为(0.61199(8)a* + 0.5c*)。最后,Pb和Bi的占有率和位置的调制参数通过同步辐射PXRD和反常散射PXRD精修得到。
8 结论
本文总结了X射线衍射晶体学和电子晶体学在复杂无机晶体结构解析中的应用,针对粉末X射线衍射峰重叠,样品不纯,骨架结构存在无序,客体分子存在无序,赝对称性,非周期性结构等常见问题怎样灵活运用X射线晶体学和电子晶体学技术完成结构解析。电子晶体学已经被证明是一种确定纳米尺寸晶体结构的独特方法,与X射线粉末衍射不同的是,纳米尺寸单晶的电子衍射没有峰重叠的问题,仅使用电子衍射数据也可以确定晶体的原子结构。近十年来,3D电子衍射技术发展使得利用电子衍射进行结构解析十分有前景,数据分辨率能够达到亚埃级别。高速照相机,冷冻样品台和数据收集方法的最新进展使得电子束敏感的纳米晶样品也能快速得到高质量的3D电子衍射数据并在短时间内完成结构的解析56-59。近几年,利用电子衍射技术解析了大量的球蛋白结构60,小分子蛋白结构61,多肽结构62,无机/有机化合物63,64。由于动力学效应和电子辐照损伤的影响,结构精修还主要依赖于X射线衍射数据,电子晶体学目前仍然只是X射线衍射的补充方法。在电子晶体学成为标准的结构测定方法前,仍然有很多重要的问题需要解决。可喜的是,将电子衍射数据的动力学效应考虑进去的精修方法正在发展中,利用动力学效应精修,电子衍射数据能直接观察到H原子65,甚至解析手性药物纳米晶体的绝对构型66,67。因此,我们有理由相信不久的将来,电子晶体学作为标准的结构解析方法将很快成为现实。
猜你喜欢
杂志排行

物理化学学报的其它文章
- Vapor-Liquid-Solid Growth of Bi2O2Se Nanoribbons for High-Performance Transistors
- 四卤化锰(II)配合物的结构调控及力致发光性能
- Conformational Switching of Verdazyl Radicals on Au(111)
- Photocatalytic CO2 Reduction Using Ni2P Nanosheets
- Swelling Characteristics of g-C3N4 as Base Catalyst in Liquid-Phase Reaction
- 聚合物支撑的金属有机骨架膜的制备及其气体分离性能