水铁矿结构稳定性及对砷固定研究与展望
2020-03-25杨忠兰曾希柏孙本华白玲玉苏世鸣王亚男吴翠霞
杨忠兰,曾希柏,孙本华,白玲玉,苏世鸣,王亚男,吴翠霞
(1.中国农业科学院农业环境与可持续发展研究所/农业农村部农业环境重点实验室,北京100081;2.西北农林科技大学资源环境学院,陕西 杨凌712100)
水铁矿为具有亚稳态结构、粒径小(2~6 nm)、比表面大(约610 m2·g-1)及低纯质子零点电荷(8.0~8.7)的纳米铁氧化物[1],其在环境中具有许多化学反应进程,如对砷的吸附/解析、沉淀/共沉淀及矿物转化等[2],是固定砷元素最常用的钝化剂[3]。铁循环过程控制着土壤中铬、砷、锑等变价金属元素的环境行为,并间接影响土壤中稳定价态金属元素的移动[4]。但由于水铁矿结构不稳定,易受环境因素的影响而转化为赤铁矿、针铁矿、纤铁矿等衍生矿物[5],从而影响其对砷的固持等环境行为。为此,本文从水铁矿结构、影响水铁矿结构稳定和对砷固定因素等进行了概述,并以此为基础提出了进一步研究的重点,为有效利用水铁矿降低土壤/水体中砷的活性提供参考。
1 水铁矿的结构
水铁矿是Fe3+水解过程中最先出现的一种沉淀物,也可通过存在结晶抑制剂(有机物质、硅酸根、磷酸根等)时Fe2+快速氧化形成[6]。水铁矿的结构不仅与陆地和水生系统中的离子络合相关[7],而且与自然界中矿物的转化、有机体中的铁储存及温室气体的释放等生物地球化学过程高度相关[8]。近年来,利用各种新技术对水铁矿的结构进行了深入研究,其主体结构也越来越清晰。
1.1 水铁矿的结构模型
水铁矿的结构最早是由Drits等[9]提出的三相模型,即六方单元晶胞参数为α=296Å、c=9.4Å的无缺陷f相;c=9.4Å的有缺陷d相;25%相干散射尺寸为10~20Å的高度分散赤铁矿结晶相。Michel等[10-11]利用X射线总散射配对分布函数表明水铁矿为单相物质,结构中包含20%四面体配位Fe和δ-Keggin区域(图1a)。Hiemstra[5]根据Michel的模型提出表面损耗模型,即能评估水铁矿表面反应位点的类型和数量。这两个模型是目前的主流观点。水铁矿结构的主体(75%的铁)为八面体配位,表面是四面体配位[12-13]。八面体是基于阴离子形成的面状结构(平均距离为0.23~0.25 nm)在晶体特殊方向上堆积而成。铁原子存在于面状结构的空隙中,被6个O+OH以八面体(γ型)或六面体(α型)的形式紧密结合在一起,使结构中的电子达到平衡,并根据排列顺序形成不同的铁氧化物[14]。不饱和的四面体是水铁矿的转化和吸附位点,导致水铁矿具有高的吸附性能和表面活性[8]。水铁矿可以在保持矿物结构稳定的情况下脱水,达到OH/Fe仅为0.2的理想结构[Fe10O14(OH)2][15]。水铁矿颗粒中O和H约145、252个,由于表面基团的形成,仅有40%的氧存在完全配位;氢的含量相当于质量为16.2%的H2O(g/100 g水铁矿纳米颗粒),其中与结构OH有关的氢含量仅相当于1.9%的H2O。尽管水铁矿颗粒中富含H2O,但矿物核心贫氢且多面体数量较少,使其具有表面缺陷的特征;水铁矿平均粒径(d)为2.58 nm,而核心结构的粒径为2/3 d[5]。这个结果与Drits等[9]提出的水铁矿核心具有大量缺陷和水铁矿结构中有丰富的OH的结论相悖。
1.2 水铁矿结构中的铁
基于 Hiemstra[5]和 Michel等[11]的结构模型,水铁矿结构中铁总数约为215个[5,8],由3种铁(Fe1、Fe2、Fe3)组成:前两种Fe(含量分别为60%和20%)为六配位,Fe3(20%)为四面体配位[5],且Fe2和Fe3的含量随着水铁矿粒径的增大而增加。在水铁矿表面,两个Fe1通过共边连接,以行的形式形成单配位表面基团(图1c),而3个Fe2和一个Fe3将Fe1固定在中间组成水铁矿的结构[12]。Fe2具有3条短(1.952Å)和3条长(2.21Å)的Fe-O键(图1b)[11],造成水铁矿表面发生强烈扭曲[16]和结构的不稳定性。同位素57Fe标记水铁矿老化10~80 d后显示约26%的Fe是不稳定的,且这些不稳定的Fe与Fe1相关。利用穆斯堡尔谱对水铁矿精细拟合,结果表明该不稳定位点具有较大的四极距分裂,这是由于水铁矿相邻的八面体Fe数目较少,使部分不稳定的Fe更加无序[5,15]。
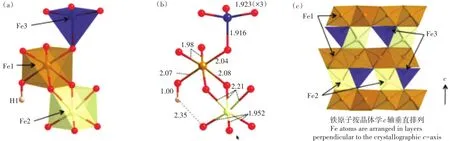
图1 密度泛函理论优化的水铁矿结构(a);水铁矿结构中部分键长(Å)(b);Fe1和混合的Fe2/Fe3的分层排列结构(c)[16]Figure 1 DFT-optimized ferrihydrite structure(a);Calculated partial bond lengths(Å)in ferrihydrite are shown(b);Part shows the layering arrangement of Fe1 and mixed Fe2/Fe3 layers(c)[16]
1.3 水铁矿结构中的OH和H2O
水铁矿的弱结晶结构与大量的铁和氧位点被OH/H2O取代有关。水铁矿结构包括2种分布在1~11和1~10面的单配位氧(占水铁矿表面积的约75%),和5种分布在001面和00-1面的三配位氧[1]。水铁矿的不稳定性主要体现在H2O和OH的行为:在一定条件下,通过微弱的物理、化学作用吸附在水铁矿[FeO(OH,H2O)a]表面的H2O先脱水聚合形成FeO(OH,O)a,接着失去OH产生空位来维持矿物电荷的平衡,最后转化为赤铁矿(Fe2On)[2]。
通过热重分析可知水铁矿质量的损失可以分为3种:(1)低温加热时,损失以物理作用吸附在水铁矿表面的 H2O[12,14];(2)温度高于 125 ℃时,损失以化学作用吸附的、“≡OH”和“≡OH2”表面基团形式存在的H2O,且水铁矿核心的结构OH也会有部分损失[12];(3)当温度高于300℃时,水铁矿会损失OH/Fe摩尔比约为0.18的H2O[15]。对于2线(d≈2.5 nm)和6线(d≈6 nm)水铁矿,在没有物理吸附H2O的情况下,“≡OH”和“≡OH2”的含量分别相当于14%和5%的H2O,摩尔质量分别为95、86 g·mol-1Fe,相应水铁矿的化学式为Fe10O14(OH)2-n H2O,n分别为0.74(2线)和0.27(6线)[5]。
2 水铁矿结构的稳定性及其对砷的固定作用
水铁矿是一类对砷具有较强吸附能力的矿物,被广泛用作土壤中砷的改良剂。水铁矿表面的不饱和配位点能通过吸附空气中的水分子来增加平均配位数。因此As5+不仅能占据不饱和配位点,还可替代H2O解离形成的OH位点,并通过扩散键合微孔内部位点,故水铁矿表现出很高的吸附容量和OH释放量[17]。水铁矿的单配位≡FeOH(H)位点密度为(10±1)μmol·m-2、6 mmol·g-1水铁矿或 0.6 mol·mol-1Fe[5]。在结构上,被吸附的砷离子与≡FeOH(H)形成共角(2C)或共边(2E)络合物[8]。由于水铁矿表面缺陷结构,2E随着水铁矿粒径增加而迅速降低[12]。且共边位点为高能位点,在低吸附量时优先被占据。水铁矿主要通过以下方式降低砷的生物有效性:(1)矿物表面OH官能团(FeOH1/2-和FeOH21/2+)与As5+配位交换,释放表面OH2或OH进入溶液,发生质子化或络合作用[18],形成FeO6和AsO4多面体双齿共边内球表面复合物[4],而在 pH>8时存在单齿键合机制[1,8]。Manning等[19]发现铁氧化物与As5+、As3+形成双配位体结构,并在铁氧化物内表面形成As-Fe结合键;(2)部分解离的水铁矿与As5+发生共沉淀,形成更稳定的砷酸铁(FeAsO4·2H2O)[20]。
水铁矿的结构具有Fe2配位不对称和H2O/OH易丢失的特性[4,8],这导致其稳定性较差,容易转变成结晶度更高的次生矿物。在水铁矿转化过程中,氧化态As5(+HAsO-4、H2AsO24-)通过内圈交换或表面沉淀吸附在水铁矿表面,延缓水铁矿的转化速率[8],从而维持水铁矿-砷高效的稳定性[18]。As5+的浓度越高对水铁矿的抑制效果越显著:在70℃、pH 12的条件下,50%的水铁矿在8 h内转化为针铁矿或赤铁矿;当体系中增加1%mol砷酸盐时,水铁矿的转化速率延迟20倍[4];当As/Fe摩尔比从1%增加到1.8%时,水铁矿的转化率下降2个数量级[21]。从而延缓其溶解和转化过程。
水铁矿吸附As5+的稳定性仍存在争议:有研究认为水铁矿在老化过程中伴随着比表面积和吸附位点的降低、物理吸附H2O和微孔体积减小、结构OH摩尔比和介孔尺寸与体积增加而导致的Fe-As结合物中As5+的释放,但Cd、Cu和Pb的稳定性增加[12];也有研究报道水铁矿在晶型转变过程中,As5+被固定在矿物结构内部的八面体空隙中,形成稳定的化学结合键(Fe-O-As)[4]。Hu等[22]结果显示As5+可迅速吸附在水铁矿表面,随着水铁矿转化会有少部分As5+的释放,但大部分仍结合在铁氧化物晶格中,且吸附在铁氧化物中的As5+与矿物的结晶度呈正相关。结合物在培养过程中所释放的As5+与游离态Fe3+具有极好的相关性(R2=0.97),这进一步明确了土壤中铁氧化物对砷的固定作用[23]。除了水铁矿的吸附,转化矿物与砷的结合也是非常重要的稳定机制。
通常在土壤经过稳定化改良后,会使用绿色植物和相关微生物固定污染土壤根区的重金属,进而增强重金属的化学稳定性。植物覆盖显著减少土壤侵蚀和环境中污染物的渗透,并使修复场地更加美观。有研究进行了几项短期和长期的实地研究,以评估水铁矿和转化产物对污染土壤中砷和植被的影响(表1)。
3 影响水铁矿转化和对砷固定的因素
在合适的条件下,水铁矿会向热力学稳定的晶质铁氧化物转化:有氧条件下,通过溶解-再结晶或脱水-结构重排向针铁矿或赤铁矿转化[29];厌氧条件下,水铁矿转化为纤铁矿、针铁矿及赤铁矿;干湿交替条件下主要转化为赤铁矿[1,26]。总之,高温、低湿和pH接近PZC(8.7)的环境有利于水铁矿向赤铁矿转化,反之有利于向针铁矿转化[6]。水铁矿转化为晶质铁氧化物的过程分为两类:(1)组成变化的反应,包括脱水、脱羟基和氧化、还原反应;(2)结构变化的反应,包括Top转变和结构重排[30]。铁氧化物间的相互转化通常伴随着比表面积减少、反应活性降低及吸附质的生物有效性和稳定性的变化,因此矿物的转化对砷的结合与固定有重要影响。水铁矿的转化是影响铁-砷稳定的关键,需要有更多的研究来关注影响矿物结构稳定的因素,以评估其对固定污染土壤中砷的长期效率。

表1 利用水铁矿和针铁矿稳定化处理砷污染土壤的研究实例Table 1 Examples of studies dealing with arsenate stabilization in contaminated soils using ferrihydrite and goethite
3.1 温度和pH
温度显著影响水铁矿的转化速率。在1℃的条件下,水铁矿转化10%需要2年,转化50%需要10年,全部转化需要130年[4]。在4~25℃条件下,培养10年仍有水铁矿未转化[31]。在92℃的条件下,水铁矿转化25 h时Fe-O距离与赤铁矿一致[13]。在50~100℃条件下,由于水铁矿脱OH或H2O,内部结构电荷失衡而原子重排,形成更加紧密稳定的赤铁矿[6]。这些转化过程导致水铁矿的比表面积减小,随后释放被吸附的As5+。
当不存在抑制剂时,水铁矿的结晶度随时间的延长而增加,且最终转化形态取决于pH[4]。高温和中性pH有利于形成赤铁矿,低pH(2~5)和高pH(10~14)有利于针铁矿的形成。这是由于在强酸和强碱条件下Fe(OH)+2和Fe(OH)-4的含量高,质子化作用较为显著,水铁矿通过溶解-沉淀向针铁矿转化[31];在近中性条件下,由于Fe3+活性低,水铁矿形成大量Fe(OH)2+,继而脱水、原子重排而向赤铁矿转化,且该转化速率随着pH的升高或降低而降低[32]。水铁矿在25℃、pH 2条件下培养90 d,转化率约为10%,但在pH 10条件下达到该转化率仅需要21 d[4]。此外,水铁矿在50℃、pH 2条件下培养7 d出现明显的针铁矿和赤铁矿的特征峰,但在50℃、pH 10条件下培养21 h就出现上述峰强度,培养168 h转化完全。在pH 10条件下,温度从25℃升高到100℃,水铁矿的转化速率增加5个数量级[4,31],即在100℃、pH 10条件下培养3 h水铁矿转化完全[4]。
近年来关于pH影响水铁矿对不同价态砷固定能力的研究较为深入[8]:铁氧化物对As3+的吸附随着pH的增加而增加(最大pH为8~10),对As5+的吸附随pH的降低而增加(最小pH为3~5),且As5+不会在短时间内被金属或溶解铁还原。As5+为H键的受体或供体取决于体系的pH和其相邻的表面基团:在低pH条件下,As5+在水铁矿表面发生质子化或络合作用并充当H键供体,形成双齿双核表面配合物(公式1);而在中、高pH条件下,As5+在水铁矿表面发生去质子化并成为H键受体(图2),形成少量单齿配位(公式2),阻碍水铁矿的溶解和转化[8]。H键的形成有利于水铁矿在任何pH条件下保持对砷高效的吸附能力,且增加了Fe-As络合物和水铁矿结构的稳定性[33]。
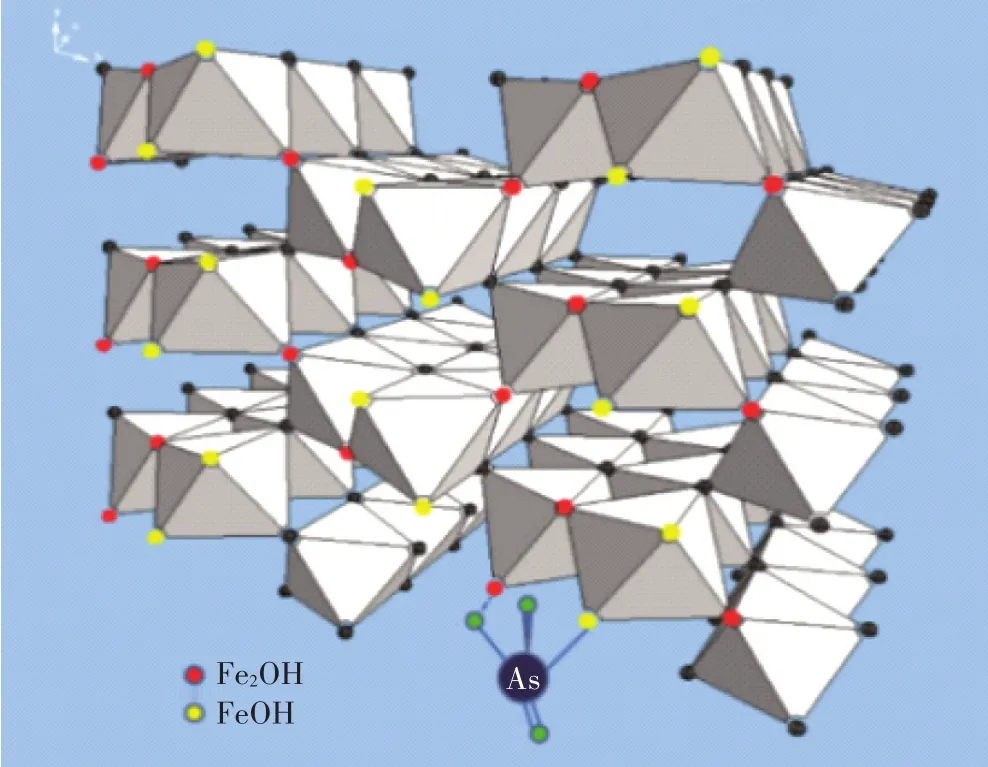
图2 模拟水铁矿(010)面和As5+形成的单和双配位表面复合物的分子结构[33]Figure 2 Molecular structure of mono-and bi-coordinate surface complexes formed by simulating ferrihydrite(010)and As5+[33]

3.2 溶液中离子的类型
离子环境是通过改变水活度而影响水铁矿的合成(表2)和转化,且高水活度有利于针铁矿的形成[34]。以硝酸盐为介质时,水铁矿通过两步结晶的方式进行转化,转化的中间产物为针铁矿,终产物以赤铁矿为主,而该过程与体系的pH和温度无关[32]。硫酸盐是专性吸附离子,更容易吸附在水铁矿的表面,取代表面吸附的As5+,并介入水铁矿的成核过程,阻碍其溶解和转化。硫酸盐介质的酸性环境有利于水铁矿向针铁矿转化,6线水铁矿为中间产物;碱性环境有利于水铁矿转化为赤铁矿[35]。这些转化过程减少水铁矿的吸附位点,且地下水和土壤溶液中常见的离子能与砷竞争吸附位点,从而影响矿物对砷的吸附。例如磷酸根与砷酸根结构相似(四面体阴离子),会竞争水铁矿表面的结合位点,并形成内圈复合物[36]。
3.3 Fe2+
Fe2+能显著提高水铁矿的转化速率,改变水铁矿的比表面积和溶解度[5],并影响负载污染物的迁移。吸附在水铁矿表面的Fe2+将电子传递到水铁矿结构态的Fe3+,导致Fe3+被还原溶解,催化水铁矿发生晶相转变[46]。水铁矿粉末加入到pH 7.2的2 mmol·L-1的FeCl2溶液培养132 h,发现水铁矿的转化率为94%,转化产物为针铁矿和纤铁矿[47]。Fe2+的浓度影响转化产物的类型[48]:在Fe2+/Fe3+摩尔比<0.02、pH<6.7时,水铁矿通过溶解-再沉淀机制转化为多晶型纤铁矿;pH>6.7时,水铁矿主要转化为针铁矿,且针铁矿的含量随着Fe2+浓度的增大而增加[49](公式3~公式6)。当0.04<Fe2+/Fe3+摩尔比<0.1时,水铁矿转化的终产物为针铁矿,其中纤铁矿为中间产物(公式7)。当0.1<Fe2+/Fe3+摩尔比<0.5时,水铁矿转化为磁铁矿[50]。
Fe2+能催化氧化铁晶相转变产生氧化能力较强的三价铁,随后化学氧化As3+为As5+[50],从而有效固定污染土壤、沉积物中的砷,但会导致土壤酸化[51](公式8)、活化金属阳离子。土壤中与Fe2+氧化或Fe3+矿物沉淀相关的微生物会影响水铁矿的结晶和结构转变[6],同时影响对砷的固定:在中性环境下,亚铁氧化菌氧化Fe2+生成水铁矿,减弱砷的移动性;而铁还原菌驱动水铁矿还原溶解导致砷的释放[52]。

3.4 有机物
在周期性干湿交替的环境中,土壤有机物通过吸附和共沉淀两种方式与水铁矿形成有机-铁复合物,从而延缓水铁矿的合成[53]并降低水铁矿-砷的稳定性。潜育土、泥炭土和水稻土由于厌氧呼吸和固相铁还原性溶解而富含Fe2+,随后氧化的Fe3+与有机物形成共沉淀[6],并非形成水铁矿,因此Fe2+的氧化和与有机物的共沉淀对吸附过程占主导[54]。
有机物能阻碍水铁矿的溶解聚合过程,影响水铁矿的粒径、比表面积、晶格结构及矿物组成[50]。体系中存在单糖时,水铁矿转化的主要产物为赤铁矿,其影响顺序为麦芽糖>葡萄糖>蔗糖[11]。羟基羧酸抑制水铁矿转化的能力为柠檬酸>内消旋酒石酸>L-酒石酸[55]。体系中存在高浓度醋酸盐时,水铁矿主要转化为针铁矿,低浓度时主要转化为赤铁矿。半胱氨酸能加快水铁矿的转化速率,且改变转化产物的类型[56]:在pH 6、70℃条件下,铁/半胱氨酸摩尔比为10时形成针铁矿和赤铁矿的混合物,比例为5时主要形成纤铁矿,比例为2.5时只形成针铁矿。研究发现当Fe2+浓度为0.2 mmol·L-1时,有机物促进水铁矿转化为纤铁矿并抑制针铁矿和磁铁矿的生成;当Fe2+浓度为2 mmol·L-1,有机物促进水铁矿转化为针铁矿[54]。随着有机物含量的增加,抑制水铁矿转化为晶质铁氧化物的能力增强。当体系中C/Fe摩尔比高于1.6时将完全抑制水铁矿的转化。这是由于有机物吸附在水铁矿表面位点并聚集,阻碍了水铁矿电子穿梭和矿物溶解[57]。同时有机酸主要通过有机配位体交换(公式9)和配位体诱导使水铁矿溶解(公式10)[58]并产生Fe3+胶体[59],从而降低矿物对砷的固定能力。
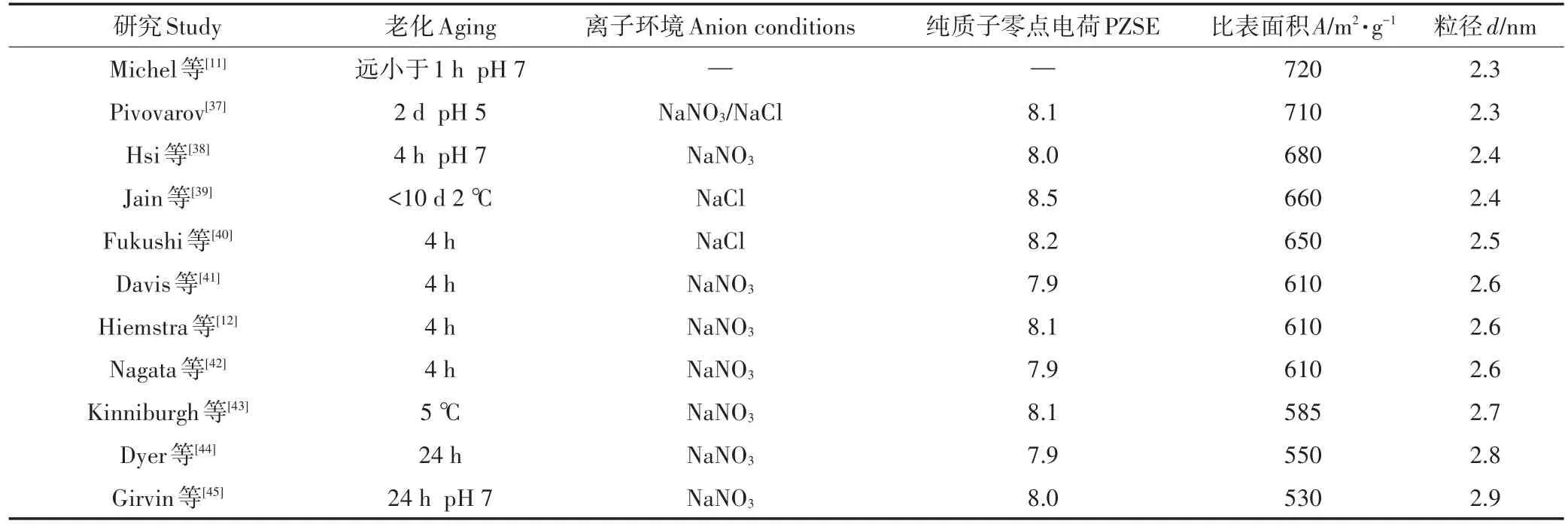
表2 不同离子环境下制备的水铁矿的比表面积和粒径Table 2 Specific surface and particle size of ferrihydrite prepared under different anion conditions

式中:FhyOH为水铁矿;L为有机酸;Asaq为游离态砷。
3.5 土壤矿物
土壤矿物能显著影响水铁矿的转化速率和产物类型[60]。自然界中,Si促进Fe2+优先氧化为水铁矿,且水铁矿的部分不饱和位点被Si取代形成Fe-O-Si键(通常Si/Fe摩尔比为0.1~0.4)[4],阻碍水铁矿的聚集和转化。随着Si含量的增加,Si对水铁矿转化为针铁矿的抑制能力大于赤铁矿[10]。这是由于针铁矿和赤铁矿的结晶表观活化能随着Si含量的增加而增加,但针铁矿增加得更急剧。水铁矿与Si单聚体键合形成不带电的单齿复合物,其外部OH通过H键(图3a中蓝键)与相邻的≡FeOH相互作用,导致电荷转移,阻碍≡FeOH的质子化及Si-OH配体的去质子化,形成稳定的Fe-Si结合物[≡FeOHFeOSi(OH)3][12]。而两个四面体Si与两个八面体Fe键合,形成稳定的双齿单核复合物(图3b和图3c),延缓水铁矿的转化。结合物的结构与Si单体相似:附着在Fe八面体上的Si四面体的 Si-Si距离(301 pm)和 Si四面体的 Si-O-Si角(128°)接近于斜方辉石的值(305 pm和131°)[5,8]。
土壤组分影响水铁矿的转化,其抑制顺序为黏土>高岭石>三水铝石>石英,而白云母加快其转化[39]。除黏土外,其他土壤组分均有利于水铁矿向赤铁矿转化,这可能是由于模板效应和黏土矿物溶解释放少量铝的作用[40]。当水铁矿在pH 5、25℃且存在土壤矿物下老化8.4年,三水铝石、高岭石、伊利石和蒙脱石延缓水铁矿的转化,而水铝英石和含蒙脱石的黏土几乎完全抑制水铁矿的转化[31]。尽管土壤组分与水铁矿的表面络合作用阻止矿物的重结晶,并保持对砷高效的吸附能力,但硅酸根为四面体阴离子,能与As5+竞争水铁矿表面的吸附位点,并形成内圈复合物[36],从而降低水铁矿-砷的稳定性。且添加过多的含铁物质会引起土壤结构的变化[61]。因此铁氧化物的使用不能只考虑钝化效果,添加量、高费用及次级污染物对土壤结构的影响[62]也是需要重视的问题。
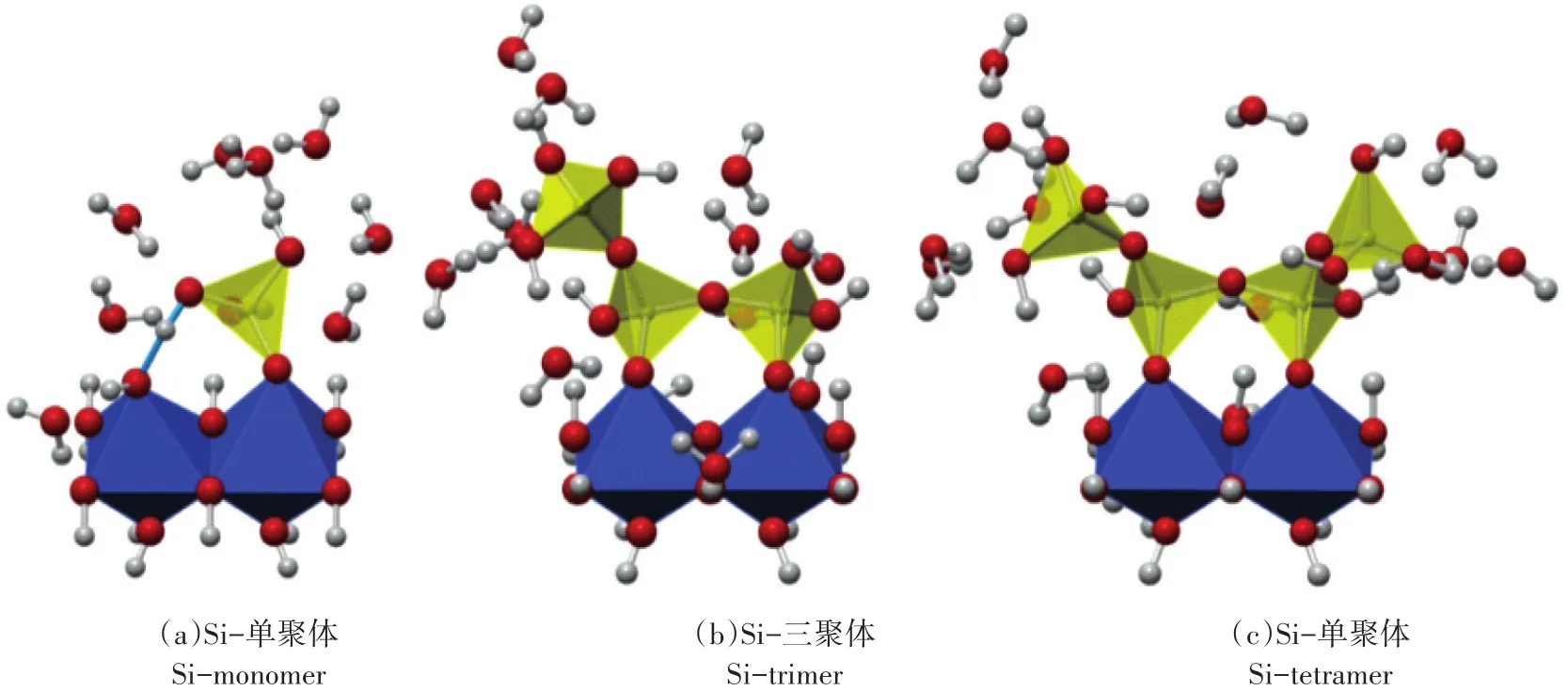
图3 3种代表性Fe-Si复合物的结构模型[12]Figure 3 The geometries structures of the three representative Fe-Sicomplexes[12]
4 问题和展望
根据现有研究结果及进展,围绕提高水铁矿对砷的固定能力、保证水铁矿-砷结合物的稳定性等,仍需强化以下研究:
(1)基于盆栽或农田水分管理、种植制度调整等技术,确定水铁矿的最优添加量,最大程度降低水铁矿对环境的影响,深入探讨水铁矿对农田中砷的固定过程;除了效率评估,修复剂的成本和当地的经济可行性也需要关注。含铁物质在土壤介质中的行为、归趋和生态系统风险也需科学评价。
(2)基于光谱表征技术,明确土壤中水铁矿-砷结合物的稳定性机制,建立可靠模型,最终实现通过机理模型对水铁矿在不同环境条件下转化和固定砷的有效预测。
(3)基于多组分、多污染离子条件下,需要密切评估砷在天然铁氧化物表面吸附过程中,对高等植物、根区分泌物、土壤生物群和人类的影响,阐明复杂环境中As-Fe离子的交互作用。
(4)基于室内研究土壤中含铁物质的地球化学变化导致重金属再活化和增加植物吸收的结果,需要进行更长期的田间试验来评估其稳定性、重金属迁移率和植物吸收、植被恢复强度及植物稳定效率。
