胶束电动毛细管色谱-激光诱导荧光检测法测定番茄酱、番茄沙司和辣椒粉中罗丹明B
2020-03-11丁晓静刘文叶
丁晓静,刘文叶,王 萍
(1.北京市疾病预防控制中心 食物中毒诊断溯源技术北京市重点实验室,北京 100013;2.北京市预防医学研究中心,北京 100013;3.首都医科大学公共卫生学院,北京 100069)
罗丹明B是一种碱性荧光染料,经常用作荧光标记物用于临床和食品检测[1-2],因其易于大量合成,具有成本低廉、色泽鲜艳且着色能力强、性质稳定不易脱色的特点,也曾经用作食品添加剂,但后来实验研究证明罗丹明B对人体有害[3],不允许在食品中添加。我国出入境检验检疫行业标准中制定了进出口食品中罗丹明B的高效液相色谱(high performance liquid chromatography,HPLC)检测以及HPLC-串联质谱(HPLC-tandem mass spectrometry,HPLC-MS/MS)检测和确证方法[4],检出限均为5 μg/kg。我国农业标准中制定了饲料中罗丹明B的HPLC法,检出限为1 μg/kg[5]。建立简单、快速而高灵敏度的测定食品中罗丹明B的分析方法,对监管食品安全具有重要意义。
目前罗丹明B的检测方法有HPLC[6-10]、超高效液相色谱-高分辨质谱(ultra-high performance liquid chromatography-high resolution mass spectrometry,UPLC-HRMS)法[11]、UPLC-MS/MS法[12-15]、毛细管电泳(capillary electrophoresis,CE)法[16-21]、CE-MS法[22]、分光光度法[23-24]和表面增强拉曼光谱(surface-enhanced Raman scattering,SERS)法[25-26]。HPLC及UPLC-MS/MS法因具有广泛的适用性而成为痕量分析的主流检测技术,但其复杂耗时的前处理、大量的有机试剂消耗而导致其检测成本高;分光光度法因无法避免样品基质的干扰而易导致定性及定量准确性欠佳;SERS法尽管快速且涵盖的样品种类多,但该法更适合于筛查或实验室预检。
CE除分离效率高、检测成本低之外,其样品前处理过程也比HPLC更为简单、有效和快捷,可满足基体复杂的不同食品分析要求,已用于工业染料的检测[17,20]。而用激光诱导荧光(l a s e r-i n d u c e d fluorescence,LIF)进行检测,可获得更高的检测灵敏度[19],非常利于较少添加量即可达到着色效果的样品检测,然而番茄酱、番茄沙司和辣椒粉中罗丹明B的CE-LIF分析方法鲜见文献报道。本研究建立罗丹明B的胶束电动毛细管色谱(micellar electrokinetic capillary chromatography,MEKC)-LIF分析方法,优化与分离缓冲溶液相匹配的样品前处理方法,成功实现了番茄酱、番茄沙司和辣椒粉样品的分析。其中实验室自制的3 个番茄酱和3 个番茄沙司的质控参考样的检测结果与本实验室建立的UPLC-FLR法[27]相比较,取得了较为理想的结果。
1 材料与方法
1.1 材料与试剂
辣椒粉 市购;番茄酱和番茄沙司质控阳性参考样的制备:市购并经本实验室用UPLC-MS/MS检测确认为空白样品,将此空白样品加标并充分混匀制得。
未涂层熔融石英毛细管(内径75 μm) 河北邯郸永年光纤色谱有限责任公司。
十水合四硼酸钠(Na2B4O7·10H2O,纯度≥99.5%) 中国医药公司北京采购供应站;十二烷基硫酸钠(sodium sodecyl sulfonate,SDS,纯度≥99%)美国Sigma-Aldrich公司;脱氧胆酸钠(sodium deoxycholate,SD,纯度≥98%) 北京百灵威科技有限公司;聚乙二醇35000(polyethyleneglycol,PEG 35000) 美国Fluka公司;NaOH(优级纯) 北京化工厂;罗丹明B(纯度≥99.0%) 德国Dr. Ehrenstorfer GmbH公司。
标准储备液:准确称取标准品罗丹明B 10.0 mg于预先装有5 mL超纯水的10 mL棕色容量瓶中,待溶解、稀释后,再加入超纯水定容至刻度,涡旋混匀后,得到质量浓度为1.0 g/L的标准储备液,于4 ℃冰箱避光保存。
1.2 仪器与设备
Beckman PA 800 plus型CE仪(配LIF检测器)美国Beckman公司;KQ-100E医用超声波清洗器 江苏昆山市超声仪器有限公司;Milli-Elix/RiOs超纯水仪美国Millipore公司;F-50A酸度计 北京屹源电子仪器科技公司;Universal 32高速离心机 德国Hettich公司;Vortex-Genie 2涡旋混合器 美国Scientific Industries公司;分析用研磨机 德国IKA公司。
1.3 方法
1.3.1 CE测定条件
未涂层熔融石英毛细管:75 μmh30 cm(有效长度20 cm),分离缓冲溶液:30 mmol/L Na2B4O7+20 mmol/L SDS+20 mmol/L SD+0.2 g/L PEG 35000;样品提取液:10 mmol/L SDS。样品室温度15 ℃,操作温度25 ℃,分离电压8 kV,工作电流65 μA左右,进样压力3.448 kPa,进样时间5 s;激发波长488 nm,发射波长520 nm。
1.3.2 毛细管的预处理
新装毛细管在使用前分别用1 mol/L NaOH溶液冲洗20 min;超纯水和分离缓冲液均冲洗5 min。每次进样前依次用1 mol/L NaOH溶液、超纯水和分离缓冲液均冲洗2 min,从而保证结果的可靠性。
1.3.3 样品前处理
辣椒粉:称取0.3 g,置于15 mL离心管中,加入样品提取溶液10 mmol/L SDS至10 mL刻度,涡旋2 min后离心5 min,取出上清液,至另一50 mL离心管中,再根据上述过程重复提取一次,合并2 次提取液,加入样品提取溶液至20 mL刻度,根据样品中罗丹明B的响应值,用样品提取液将其稀释适当倍数直接进样。
番茄酱和番茄沙司:称取0.3 g,置于15 mL离心管中,加入样品提取液10 mmol/L SDS至10 mL刻度,涡旋混匀后离心5 min,取上清液直接进样即可。
1.4 数据统计
使用SPSS 19.0软件进行数据统计,CE图均由32 Karat 8.0软件直接输出,表格用WPS 2019办公软件绘制而成。
2 结果与分析
2.1 实验条件的选择
2.1.1 分离缓冲体系的选择
当pH值低于3时,罗丹明B完全以阳离子形式存在,而当pH≥8时,以中性分子形式存在[21,28-29]。有研究用硼砂缓冲体系作为分离缓冲溶液,可获得很好的分离效果[19,27]。也有用磷酸盐体系作为分离缓冲溶液的报道[18],但磷酸盐因电导值较高,浓度过高会产生较多的焦耳热,造成峰展宽,分离效率下降;浓度太低则使得待测物与样品基质不能很好分离。故本研究选用硼砂缓冲体系。
2.1.2 分离缓冲溶液中盐浓度的优化
通过增加分离缓冲溶液中的盐浓度,以增加离子强度而减少罗丹明B在毛细管内壁的吸附。保持分离缓冲溶液中20 mmol/L SD、20 mmol/L SDS和0.2 g/L PEG 35000不变,考察不同浓度(20、30、40 mmol/L)硼砂对罗丹明B标准溶液出峰的影响,结果见图1。硼砂浓度对罗丹明B的检测灵敏度没有明显影响,然而随着硼砂浓度的增加,将引起峰的稍微展宽,导致迁移时间相应增加。为保证与样品基质能得到充分分离,选择30 mmol/L硼砂为最佳。
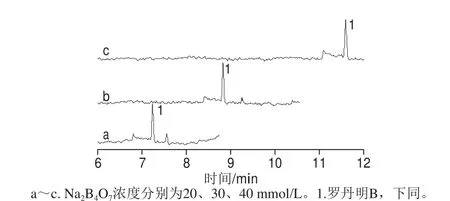
图1 分离缓冲溶液中硼砂浓度对罗丹明B峰形及迁移时间的影响Fig. 1 Effects of Na2B4O7 concentration on the migration time and peak shape of rhodamine B
2.1.3 分离缓冲溶液pH值的选择
分离缓冲溶液的pH值不仅可以影响毛细管壁的质子化程度以及罗丹明B的存在形式,也控制电渗流(electroosmosis flow,EOF)的大小,影响分析物的迁移时间和分离效率。分离缓冲溶液的pH值越接近硼酸的解离常数(pKa9.24),其缓冲能力也越大,可获得良好的定性及定量重现性。无论用NaOH还是CsOH调节pH值,都将增加盐浓度,导致分离时间延长,罗丹明B的峰形变差。而不加入任何调节剂时,30 mmol/L Na2B4O7的pH值为9.21,接近硼酸的解离常数(pKa9.24),能够获得较好的分离效率。故本研究选用30 mmol/L Na2B4O7即可,无需调节pH值。
2.1.4 分离缓冲溶液中SDS浓度的优化
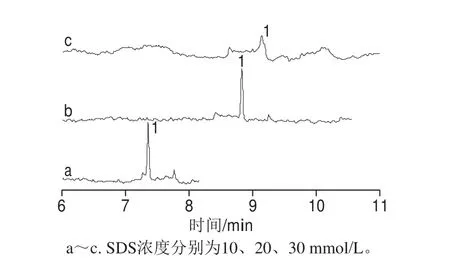
图2 分离缓冲溶液中SDS浓度对罗丹明B分离及峰形的影响Fig. 2 Effects of SDS concentration on the migration time and peak shape of rhodamine B
当pH≥8时,罗丹明B以中性分子形式存在,故采用非常适合中性分子分析的MEKC分离模式,即向分离缓冲溶液中加入能够形成胶束相浓度的SDS,在促进罗丹明B溶解的同时,还能增加分离。在碱性分离缓冲体系中SDS带负电,分离时其运动方向与EOF方向相反,但由于在碱性条件下,EOF速率大于SDS的迁移速率,包裹中性分子罗丹明B的SDS可以被EOF推至检测窗口而被检测到。25 ℃的操作温度下,能形成胶束的SDS浓度是8.1 mmol/L[30],所以考察了10、20 mmol/L和30 mmol/L SDS对分离的影响。保持分离缓冲溶液中30 mmol/L Na2B4O7、20 mmol/L SD以及0.2 g/L PEG 35000保持不变,当SDS为10 mmol/L时,罗丹明B与未知峰的分离效果不好,未达到基线分离;当SDS溶液浓度增至30 mmol/L时,罗丹明B峰变得畸形,而且灵敏度大大降低;而当SDS浓度为20 mmol/L时,罗丹明B灵敏度和分离度最好,且达基线分离。因此20 mmol/L SDS为最佳,结果如图2所示。
2.1.5 分离缓冲溶液中SD浓度的优化
SD是常用阴离子表面活性剂,含有疏水甲基和亲水羧酸根,具有较强的增溶性和聚合性,使其分离能力优于SDS[31]。保持30 mmol/L Na2B4O7、20 mmol/L SDS及0.2 g/L PEG 35000浓度不变,考察SD浓度分别为10、20 mmol/L和30 mmol/L时,对罗丹明B的分离及峰形的影响。随着SD浓度的增加,罗丹明B的灵敏度下降;当SD为10 mmol/L时,罗丹明B的灵敏度最高、峰形也好,但用于实际样品分析时,罗丹明B与样品基质无法实现完全分离;当SD浓度为20 mmol/L时,虽然罗丹明B的灵敏度较10 mmol/L时略有降低,但样品分离达到最好,故选择20 mmol/L SD,如图3所示。
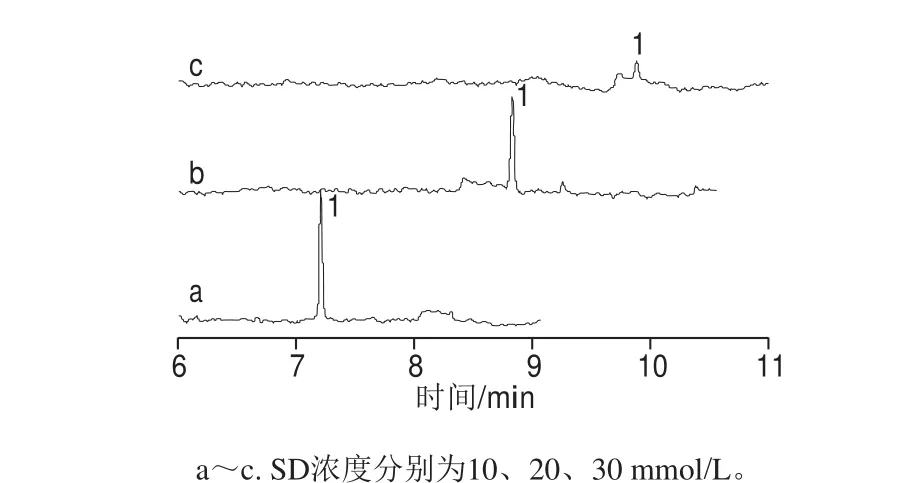
图3 分离缓冲溶液中SD浓度对罗丹明B灵敏度的影响Fig. 3 Effects of SD concentrations on the detection sensitivity of rhodamine B
2.1.6 分离缓冲溶液中添加剂及浓度的选择
PEG 35000在毛细管中起动态涂层的作用,可以减少管壁对待测物的吸附,改善峰拖尾。保持30 mmol/L Na2B4O7、20 mmol/L SDS以及20 mmol/L SD不变,未加入添加剂时,在10 min内未检测到罗丹明B的峰,加入0.2 g/L PEG 35000时,罗丹明B的灵敏度高、分离好,且用时短,如图4所示,当加入0.4 g/L PEG 35000后,罗丹明B的峰形变差,灵敏度降低。因此选用0.2 g/L PEG 35000作为添加剂。
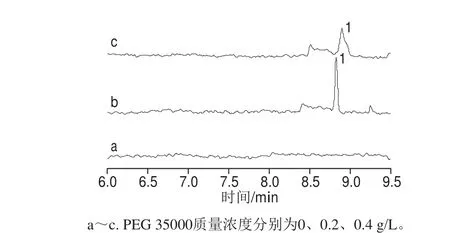
图4 分离缓冲溶液中PEG 35000质量浓度对罗丹明B检测灵敏度的影响Fig. 4 Effects of PEG 35000 concentration on the detection sensitivity of rhodamine B
2.1.7 分离电压的选择
分离电压的高低直接影响着待测物的迁移时间,电压越高,迁移速率越快,检测到待测物的时间越短,与此同时焦耳热也增加,导致罗丹明B的峰区带展宽,但是分离电压过低导致迁移时间延长,经优化,本实验采用8 kV电压进行分离。
2.1.8 进样时间的选择
分别对比了2、5 s和8 s进样时间对分离效果的影响,随着进样时间的延长,达到8 s时,虽然罗丹明B的峰高有明显增加,但出现峰分叉现象,或是平头峰;而当进样时间为2 s时,峰高降低,不利于样品中低含量罗丹明B的检测;进样时间为5 s时,灵敏度和峰形达到最优,最终选择进样时间为5 s。
2.1.9 样品提取溶液的选择
分别用超纯水、稀释10 倍后的分离缓冲液、10 mmol/L SDS提取样品,当用纯水和稀释10 倍后的分离缓冲液作为样品提取液时,罗丹明B的峰不仅灵敏度低,峰形还展宽变差,当用10 mmol/L SDS时,罗丹明B的峰变锐变高,有明显的增敏作用。又考察5 mmol/L和15 mmol/L SDS对样品的提取效果,发现两者灵敏度和分离度都不如10 mmol/L SDS合适,故选择10 mmol/L SDS作为样品提取液。
2.2 标准曲线及检出限
将1 g/L罗丹明B储备液用样品稀释液稀释,配制成质量浓度分别为0.02、0.04、0.08、0.16、0.32、0.64、1.28 mg/L的标准系列,在建立的最佳条件下,由低质量浓度到高质量浓度依次进样检测,用校正峰面积(峰面积除以迁移时间)外标法定量,以CE峰的校正峰面积(A)为纵坐标,与其对应的质量浓度ρ(mg/L)为横坐标,绘制标准曲线。其线性回归方程为A=2 489.41ρ+11.72,线性范围为0.02~1.28 mg/L,相关系数为0.999 9,检出限及定量限分别为5 μg/L和15 μg/L。
2.3 仪器精密度
分别在0.04、0.16 mg/L及0.64 mg/L三个不同质量浓度水平测定仪器精密度。迁移时间的精密度分别为0.25%、0.28%和0.10%(n=7);相应校正峰面积的相对标准偏差(relative standard deviation,RSD)分别为3.6%、1.5%和3.0%。
2.4 方法精密度
根据1.3.3节样品前处理方法处理番茄沙司和辣椒粉样品,分别平行处理7 份,在最佳MEKC条件下进行测定,计算样品含量的RSD,得到方法日内精密度分别为2.0%和1.4%。将番茄沙司和辣椒粉样品平行处理3 份并测得平均值,连续7 d,计算7 d样品平均值的RSD,得到方法日间精密度分别为1.6%和1.8%。
2.5 加标回收率
加标回收实验的本底选择某品牌番茄酱样品,选取标曲范围内的0.04、0.16 mg/L和0.64 mg/L三个质量浓度水平进行加标。平行处理每个质量浓度的样品5 份,加入标准溶液后室温放置过夜,使加标溶液与样品基质充分混合,第2天再行提取。样品中质量浓度加标CE结果如图5所示,加标回收率分别为100.9%、101.9%和105.8%,相应RSD分别为2.0%、1.4%和3.2%。
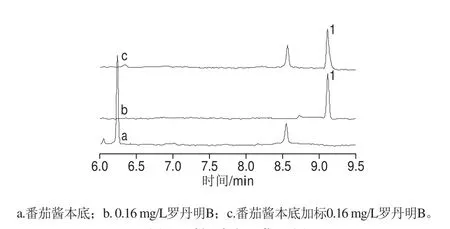
图5 样品加标回收CE图Fig. 5 Capillary electrophoregrams for spiked recovery at moderate mass concentration
2.6 样品分析结果
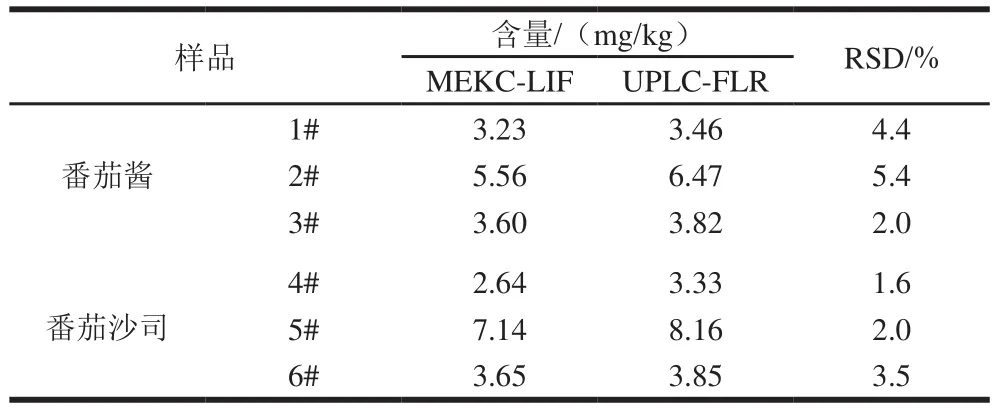
表1 番茄酱和番茄沙司实际样品分析结果(n=5)Table 1 Results of determination of rhodamine B in tomato sauce,ketchup and chili powder samples (n= 5)
按照1.3.3节的前处理方法,对3 个番茄酱、3 个番茄沙司和22 个辣椒粉共计28 个样品进行前处理,每个样品平行处理5 份进行测定,分别计算其RSD。1#~6#样品还均用UPLC-FLR的方法[27]进行了测定,测定结果见表1,经统计学检验t为0.016,小于0.05,表明两种方法的测定结果有显著性差异,可能的原因是:两种方法的检测时间相差半年,再加之样品在室温放置,故导致罗丹明B的含量有所下降,提示在今后罗丹明B的标准物质研制中,不宜将样品放置在室温,而应放置在4 ℃或-20 ℃保存。其中样品1#~3#均为番茄酱、样品4#~6#均为番茄沙司。22 个辣椒粉样品中仅有2 个未检出罗丹明B,其余20 个均检出,含量范围为13.1~798.6 mg/kg。番茄酱、番茄沙司和辣椒粉的CE图分别见图6。
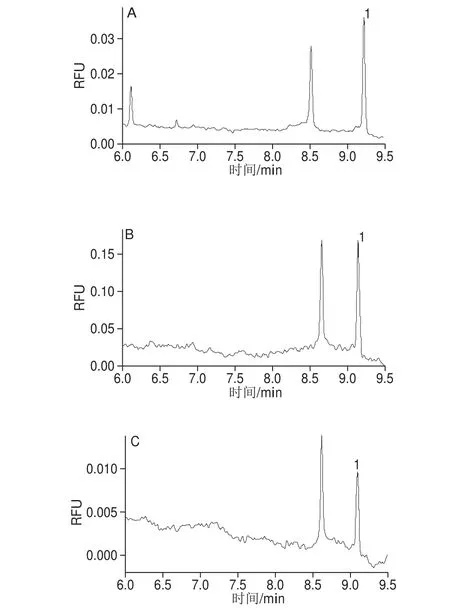
图6 1#番茄酱(A)、4#番茄沙司(B)、8#辣椒粉(C)CE图Fig. 6 Capillary electrophoregrams of tomato sauce 1# (A), tomato sauce 4# (B), and paprika 8# (C)
3 结 论
采用MEKC法,结合LIF检测器,显著提高检测灵敏度;选用硼砂盐分离缓冲体系及无需有机溶剂的简单的样品前处理,既可以提高灵敏度,又可减少前处理时间,还降低了检测成本。检出限为5 μg/L,能够满足低含量样品的检测。28 个样品中,除1#~6#样品均为本实验室制备的质控阳性参考样外,22 个辣椒粉样品中有20 个样品中均检出了罗丹明B,说明辣椒粉中非法添加的现象依然存在。尽管工业染料性质稳定,但罗丹明B的食品基体标准物质研制中,不宜将样品放置在室温,而应放置在4 ℃或-20 ℃保存。
