隐睾及隐睾合并其他泌尿生殖系统畸形的外显子测序分析
2020-02-29唐炳强耿红全林厚维徐卯升徐国锋方晓亮
唐炳强 耿红全 林厚维 徐卯升 徐国锋 方晓亮 贺 雷
隐睾是男性最常见的先天性泌尿生殖系统畸形之一,足月儿患病率为2%~4%[1],早产儿患病率高达30%[2],1岁时患病率为1%~2%。隐睾可单独发病,也可合并其他先天性畸形(如性别发育异常)[3]。目前学界普遍认为隐睾是基因、内分泌和环境相互作用并影响的结果,而基因与本病发生的因果关联仍不明晰[4]。由于胚胎性别发育的正常过程和机制尚未完全明确,且性别异常的临床表现复杂多样,因此对性发育异常进行病因诊断仍较为困难。
高通量测序技术的不断发展成熟为我们提供了新的病因学研究方法,也促进了全外显子测序技术的发展。近年来,全外显子测序作为一种快速而经济的技术已广泛应用于遗传性疾病的诊断[5]。睾丸下降过程及性别分化过程涉及的基因广泛,采用外显子测序技术可能有助于发现致病基因或新发的突变位点,为揭示隐睾和性别分化的深层次发病机制提供线索和依据。
材料与方法
一、研究对象
研究对象均就诊于上海交通大学附属新华医院,共19例。其中单纯腹股沟隐睾4例,隐睾合并尿道下裂6例,合并小阴茎6例,两性畸形2例,合并先心病1例。
二、临床资料收集
详细收集所有患儿的病史、临床表现、实验室检查及影像学检查结果,具体包括年龄、外生殖器表型(阴茎发育情况、大小、尿道开口位置、有无阴茎弯曲、睾丸的位置、大小、是否存在阴囊分裂、阴茎阴囊转位)、社会性别、家族史、染色体核型、激素水平(包括睾酮T、二氢睾酮DHT、卵泡刺激素FSH、黄体生成素LH、雌二醇E2、17α-羟孕酮、HCG激发试验)、影像学检查结果、手术史、病理结果。
三、基因测序方法
1. 基因组DNA的提取 对19例患儿外周血样本进行基因组DNA抽提(Lab-Aid核酸提取试剂盒),应用Nanodrop 2000/2000c分光光度仪检测DNA浓度及纯度(OD260/OD280比值);OD值在1.8~2.0之间、含量在1.5 μg以上的基因组DNA样品储存于-80℃冰箱中备用。
2. 建库及测序 全基因外显子测序3例,目标基因外显子测序16例。基因组DNA建库和目标区域捕获分别采用Agilent SureSelect Human All ExonV5及Inherited disease panel试剂盒。外显子区域DNA经高效富集、捕获后采用PCR线性扩增,并进行文库质检,合格的样品外显子文库通过HiSeq2500测序平台(Illumina,USA)进行高通量、高深度测序。统计各样品Illumina原始下机测序序列(Reads)的测序生成数据量、平均测序深度、覆盖度等数据。
3. 数据处理 使用BWA进行标准序列比对,对照参考序列来源于千人基因组(http://www.1000genomes.org)和美国国家生物技术信息中心第37版人类基因组序列(GRCh37),比对后得到的数据进行GATK基因型分型分析,以ANNOVAR 11注释变异,以Exome Depth检测拷贝数变异。过滤掉不影响基因功能的同义突变位点以及公共遗传突变数据库(dbSNP137数据库、千人基因组数据库、ExAC数据库)中正常人携带的常见突变位点,最后筛选出候选致病突变位点。应用SIFT、Polyphen-2及Mutation Taster预测候选基因序列变异所造成的氨基酸改变是否会导致蛋白质功能发生变化。
4.Sanger测序验证 经上述分析后筛选出可能的候选致病突变位点,通过Sanger测序方法,对患儿及其父母外周血样本进行突变位点验证,与外显子测序结果进行对比分析,并追溯突变来源。
结 果
一、患儿的基本情况
本研究共纳入19例患儿,均有隐睾表现,其中单纯隐睾4例,隐睾合并其他泌尿生殖系统畸形15例(其中2例为性别分化异常)。年龄6个月至11岁,均无家族史。
二、基因测序相关结果
1. 外显子测序结果:19例中3例为全外显子测序,16例为目标基因外显子测序。目标区域覆盖度达到98.5%以上,平均深度>10×者占比均在98%以上,提示基因组外显子捕获成功,测序质量可靠。外显子测序结果经方法3中提及的生物信息学分析过滤筛选,发现3个基因可能与相关表型发病有关: ①AR基因发生3处错义突变(c.1600C>A;p.Pro534Thr)、(c.2599G>A;p.Val867Met)和(c.528C>A;p.Ser176Arg); ②NR5A1基因移码突变(c.442delG; p.Glu148Serfs*148)和错义突变(c.43G>A;p.Val15Met); ③ATRX基因剪切位点突变(c.4317+13T>C)。结果详见表1。
2. Sanger测序结果:外显子测序经过生物信息学分析筛选得到的6处突变位点均在Sanger测序的结果中得到证实。其中5例患儿母亲存在对应位点突变,患儿父亲均未见异常,1例患儿(c.43G>A;p.Val15Met)父母均未见异常。基因序列图详见图1至图6。
三、基因型和表型的关系
AR基因突变:1例为单纯左侧腹股沟隐睾。基因测序(c.1600C>A;p.Pro534Thr);1例为性分化异常。查体:外生殖器外观与女性外阴相似,可见尿道开口及类似阴道口,双侧腹股沟区均可触及睾丸样组织。核型:46,XY。术后病理:双侧性腺均为睾丸组织。基因测序(c.2599G>A;p.Val867Met);1例表现为左侧腹股沟隐睾,阴茎短小,并有先天性巨结肠病史。基因测序(c.528C>A;p.Ser176Arg)。
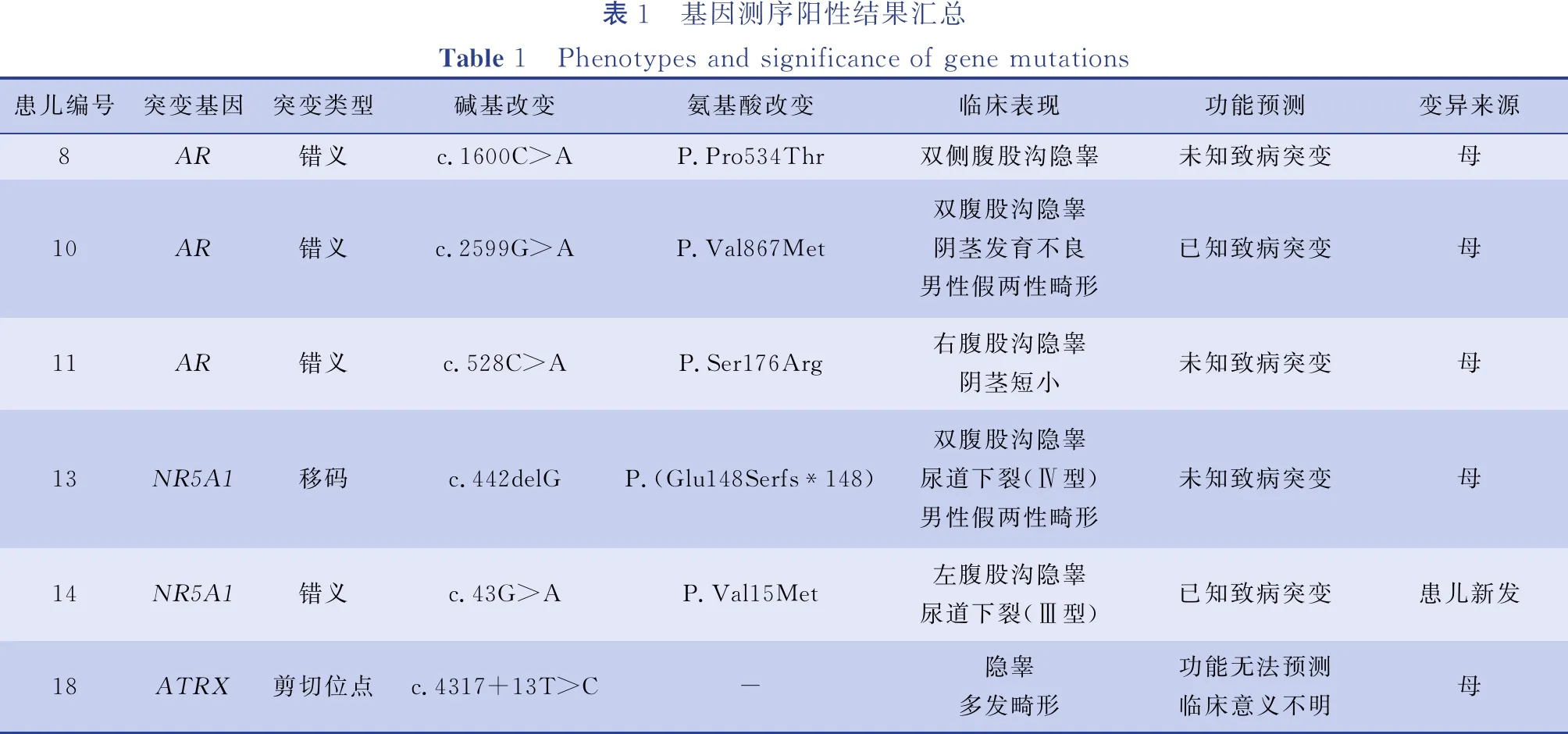
表1 基因测序阳性结果汇总Table 1 Phenotypes and significance of gene mutations患儿编号突变基因突变类型碱基改变 氨基酸改变临床表现功能预测变异来源8AR错义c.1600C>AP.Pro534Thr双侧腹股沟隐睾未知致病突变母10AR错义c.2599G>AP.Val867Met双腹股沟隐睾阴茎发育不良男性假两性畸形已知致病突变母11AR错义c.528C>AP.Ser176Arg右腹股沟隐睾阴茎短小未知致病突变母13NR5A1移码c.442delGP.(Glu148Serfs*148)双腹股沟隐睾尿道下裂(Ⅳ型)男性假两性畸形未知致病突变母14NR5A1错义c.43G>AP.Val15Met左腹股沟隐睾尿道下裂(Ⅲ型)已知致病突变患儿新发18ATRX剪切位点c.4317+13T>C-隐睾多发畸形功能无法预测临床意义不明母
图1AR基因: c.1600C>A; p.Pro534Thr (杂合,母来源)
Fig.1ARgene:c.1600C>A; p.Pro534Thr (heterozygous mutation,maternal)
图2AR基因:c.2599G>A;p.Val867Met(杂合,母来源)
Fig.2ARgene:c.2599G>A; p.Val867Met (heterozygous mutation,maternal)
图3AR基因:c.528C>A; p.Ser176Arg (杂合,母来源)
Fig.3ARgene variants:c.528C>A; p.Ser176Arg (heterozygous mutation,Maternal)
图4NR5A1基因:c.442delG;p.Glu148Serfs*148(杂合,母来源)
Fig.4NR5A1gene:c.442delG.p.Glu148Serfs*148 (heterozygous mutation,maternal)
图5NR5A1基因: c.43G>A;p.Val15Met (杂合,患儿新发)
Fig.5NR5A1gene:c.43G>A; p.Val15Met (heterozygous mutation,newly onset)
图6ATRX基因:c.4317+13T>C (杂合,母来源)
Fig.6ATRXgene:c.4317+13T>C (heterozygous mutation,maternal)
NR5A1基因突变:1例为双侧腹股沟隐睾,尿道下裂(Ⅳ型),性别分化异常。查体:外生殖器外观与女婴相似,双侧腹股沟均可触及睾丸样肿块。核型:46,XY。术后病理:双侧性腺均为睾丸组织。基因测序(c.442delG; p.Glu148Serfs*148);1例为左侧腹股沟隐睾,尿道下裂(Ⅲ型)。核型:46,XY。基因测序(c.43G>A;p.Val15Met)。
ATRX基因突变:患儿为双侧腹股沟隐睾、性腺发育不良,多发畸形(小头畸形、右上眼睑下垂、内眦赘皮、耳廓大、鼻塌、智力落后、身体发育迟缓等)。核型:46,XY。基因测序(c.4317+13T>C)。
讨 论
隐睾是常见的先天性男性泌尿生殖系统畸形之一。已有多个基因被证实与隐睾的发生有关,包括INSL3,LGR8/GREAT,HOXa10,AR,CGRP等[6],但这些基因仅可解释部分病因。性发育异常表现为染色体、性腺和附属性器官的解剖学异常,其遗传学病因复杂,且临床表型多样,正确的诊断和分类对于性别的认定、生殖器的手术治疗抉择、社会抚养性别的选择都起到非常重要的作用。
外显子组测序仅针对人类基因组序列中1%的外显子序列进行检测,具有经济、高效等特点,可更好地用于寻找致病基因和易感基因。外显子测序技术可同时检测多种基因,极大地提高了致病基因的检出率,可快速、高效地做出分子病因诊断,为临床干预提供依据。本研究通过外显子测序技术,对不同表型患儿进行基因测序分析。
雄激素是决定男性表型的重要激素,其作用主要由AR受体介导,AR基因变异会导致内外生殖器表型显著改变,甚至形成男性假两性畸形[7,8]。雄激素不敏感综合征(androgen insensitivity syndrome,AIS)是指因雄激素受体功能障碍引起的机体对雄激素出现不敏感甚至抵抗的一种疾病,而AR基因突变可导致AIS已有较为确凿的证据。目前已有超过500个关于AR基因突变的报道,这些不同的突变导致了AR基因功能出现不同程度缺失,其中约90%为单碱基突变,且绝大部分为错义突变[9]。
本研究中,1例表现为单纯腹股沟隐睾,基因测序(c.1600C>A;p.Pro534Thr);1例表现为性别发育异常(男性假两性畸形),基因测序(c.2599G>A;p.Val867Met),考虑为AIS;1例为单纯腹股沟隐睾合并阴茎短小、先天性巨结肠,基因测序(c.528C>A;p.Ser176Arg)。三种不同的突变引起了不同的临床表型。目前认为AR基因突变是AIS较为确切的病因之一,但这三种基因突变具体如何影响雄激素受体功能尚不明确,且各个突变造成的功能受限程度还有待进一步研究。
类固醇生成因子1(steroidogenic factor 1,SF1)又名核受体亚家族5组成员A1(nuclear receptor subfamily 5 group A member 1,NR5A1),是性腺和肾上腺中类固醇生成相关酶的主要调节者[10]。约有超过70个NR5A1基因突变已在多个DSD个体中被发现。在46,XY个体中,NR5A1基因参与男性性别决定和性别分化的过程,动物实验表明,基因敲除后的小鼠表现为性腺发育障碍和性发育异常,而基因测序结果也证实性发育异常患儿存在NR5A1基因突变[11,12]。
本研究中,1例表现为性发育异常(男性假两性畸形,社会性别女性),基因测序(c.442delG; p.Glu148Serfs*148),突变导致编码蛋白截短,功能预测具有致病性;1例表现为左侧腹股沟隐睾,尿道下裂(阴囊型),基因测序(c.43G>A; p.Val15Met)。该结果进一步证实NR5A1基因是生殖器官发育调节过程中的重要基因,并参与睾丸下降过程。一些杂合性突变(如p.Glu11Ter、p.Cis16Ter和p.Glu51Argfs*23)在46,XY DSD、严重阴茎阴囊型尿道下裂和隐睾患者中分别被检测出。据不完全统计,表现为不完全性腺发育不良的患者中,NR5A1基因的突变率约15%[13]。
ATR-X综合征是一种X连锁的严重智力发育障碍综合征,以易辨认的面部特征、生殖器发育异常和α-地中海贫血为主要表现。Gibbons和 Higgs报道,ATRX基因突变与泌尿生殖器发育不良存在相关性[14]。本研究中,患儿表现为多发畸形,其中泌尿生殖系统畸形主要表现为左侧腹股沟隐睾、睾丸发育不良及尿道开口异常。而ARTX基因突变个体中存在性分化的异常,表现为隐睾并睾丸萎缩和尿道开口异常。在ART-X综合征中生殖器异常的表现十分常见,可见于约80%的患者中。Aleksandra等报道,ATRX基因突变(c.G590>T,p.Cys197Phe)在1例ATR-X综合征病例中表现出生殖系统异常。本组中阳性改变患儿表现为性腺发育不良及尿道下裂,表明此基因阳性改变造成的功能改变对泌尿生殖系统发育的影响较大,其具体的作用机制仍需进一步研究。
本研究共检出3个基因的6处突变位点,其中4处为新发突变。AR、NR5A1基因突变可引起严重的性发育异常,临床表型为外生殖女性化,进一步证实46,XY DSD患者中AR、NR5A1基因突变的作用,同时ARTX基因也应引起我们的重视。
