三种5,7-二苯基-1,8-萘啶衍生物的理论光谱计算
2020-01-16张福叶徐涛迟绍明
张福叶, 徐涛, 迟绍明
(云南师范大学 化学化工学院,云南 昆明 650500)
1,8-萘啶单元存在于许多天然药物中,具有优异的生理活性,其结构中含氮原子,易与过渡金属离子和碱基对结合.因此,1,8-萘啶衍生物对各种离子的识别及在生物医学上的应用引起了研究者的广泛兴趣.2017年,Shahida U等人[1]报道了一种2-四苯乙烯-1,8-萘啶对Ag+的识别.Zhao X Z等人[2]发现1,8-萘啶衍生物可做人类免疫缺陷病毒(HIV)整合酶链转移抑制剂.2015年,Kishan Prasad C等人[3]报道了1,8-萘啶衍生物的抗菌活性.1,8-萘啶衍生物在抗癌[4]、抗炎[5-6]及抗肿瘤[7-8]等方面的作用也有研究.实验研究的同时,理论计算也不可缺,本文则运用密度泛函理论(DFT)[9-11]对本课题组合成的3种萘啶化合物进行理论计算,采用B3LYP/6-31G(d)基组优化了3种化合物的结构,并计算了优化后化合物的电子吸收光谱和前线分子轨道及能量.
1 计算方法
在密度泛函理论(DFT)方法下采用B3LYP/6-31G(d)基组优化了3种5,7-二苯基-1,8-萘啶衍生物的分子结构.进一步结合含时密度泛函理论(TD-DFT)[12]和极化连续介质模型(PCM)[13]在相同基组下计算了3种化合物在气相和液相中的电子吸收光谱,最后利用Multiwfn程序[14]和origin 95处理并绘制电子吸收光谱图及前线分子轨道和能量图.
2 结果与讨论
2.1 基态几何构型
图1展示了本文研究的3种化合物的分子结构.由图看出3种化合物的结构相似,只是具有不同取代的酰胺基团.
在B3LYP/6-31G(d)水平下优化3个化合物的基态结构的部分键长和键角数据见表1.虽然3种化合物具有不同的取代基基团,但1,8-萘啶环单元的键长和键角比较接近,部分数据可从表1 看出.由C-C和C-N的键长数据可证实原子的电负性相差越大,键长越短.
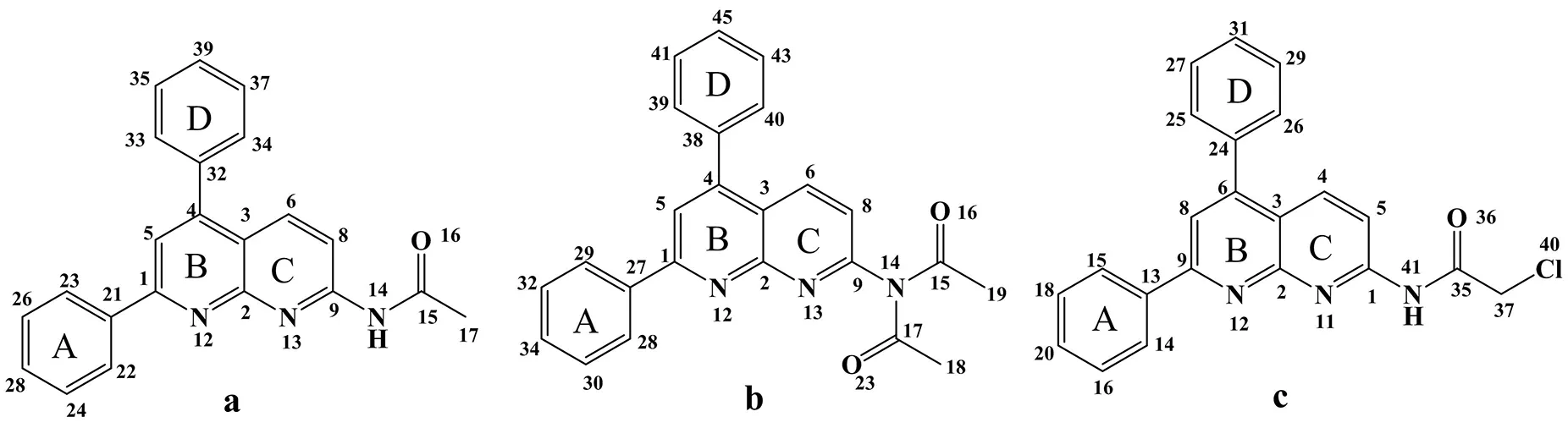
图1 3种化合物的分子结构
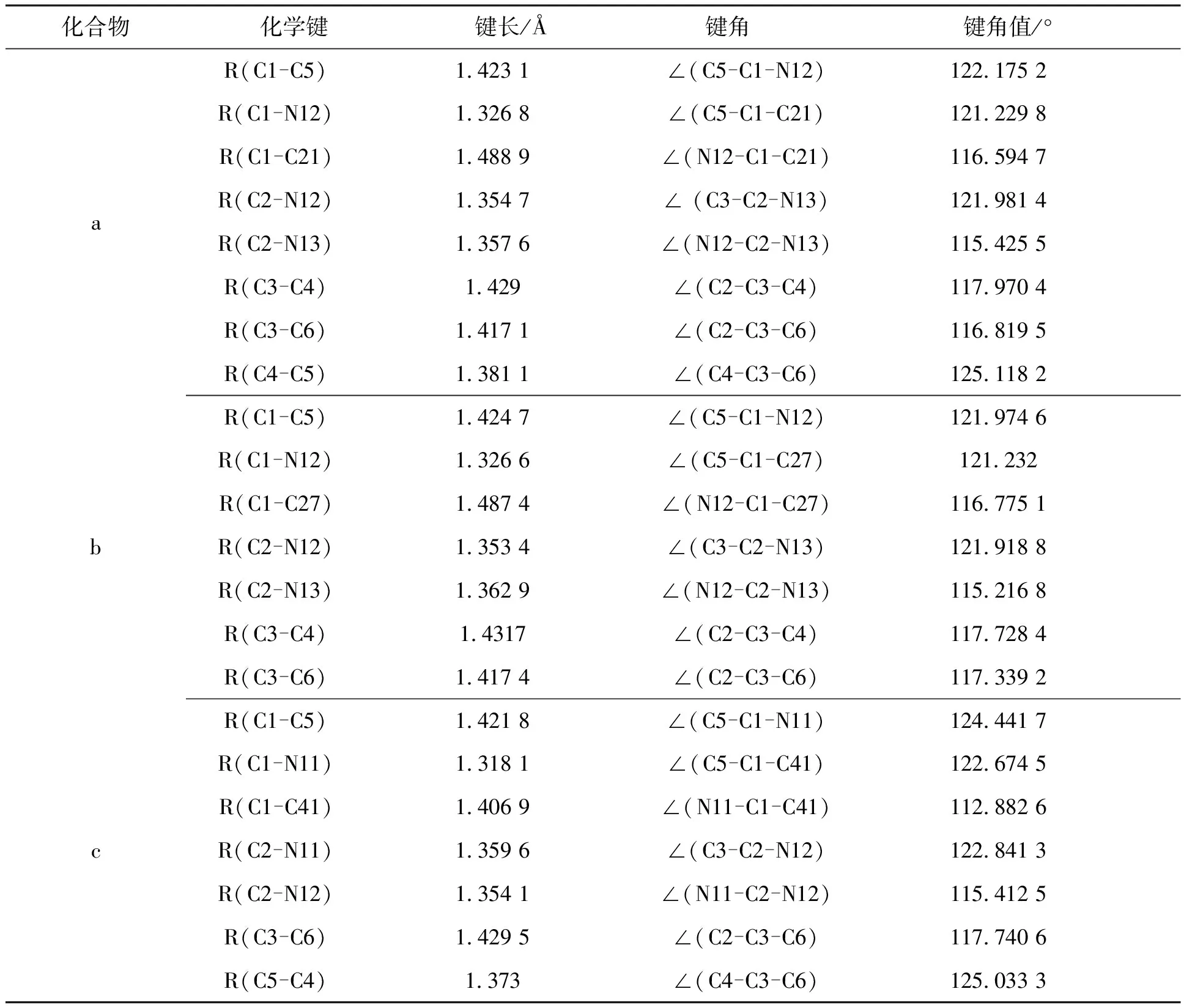
表1 3种化合物的理论计算几何参数
2.2 电子吸收光谱
在结构优化的基础上分别计算了3种化合物在气相和液相中的电子吸收光谱,得到的数据利用Multiwfn程序和origin 95处理并绘制了3种化合物的电子吸收光谱图,见图2.如图2所示,3种化合物均有2个吸收峰,260 nm前后的吸收峰起源于最高占据轨道(HOMO)的电子跃迁至最低空轨道(LUMO)+1,归属于n→π*电子跃迁,而340 nm前后的吸收起源于HOMO轨道的电子跃迁至LUMO轨道,归属于π→π*电子跃迁.
紫外可见吸收光谱在室温下由SHIMADZU UV-1780分光光度计测得,溶剂为CH2Cl2.理论最大吸收值和实验值列于表2中,从表中数据可看出3种化合物的计算值与实验值十分接近.其中3种化合物的计算值和实验值在气相中分别相差3、4 nm和22 nm,在液相中分别相差4、4 nm和19 nm,表明液相和气相的计算值与实验值差别不大,且液相值与气相值相差不大.说明这3个化合物的溶剂效应不强,与实验结果较为吻合,计算这3个化合物的电子吸收光谱采用B3LYP/6-31G(d)基组是合适的.

图2 3种化合物的电子吸收光谱图
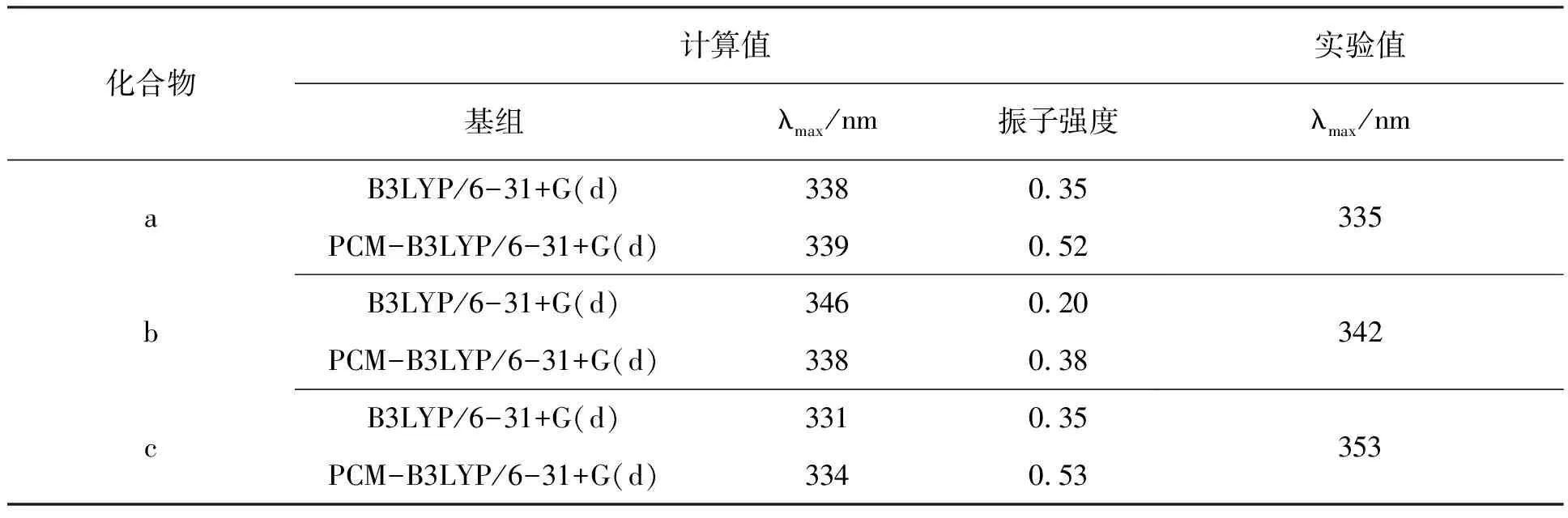
表2 3种化合物的吸收光谱计算值和实验值
2.3 前线分子轨道
图3展示了3种化合物的前线轨道及能量图,由图3可看出3种化合物的电子云分布大致相同,HOMO轨道上的电子云除了是集中在A环和萘啶环上,D环的苯取代基上无电子云分布.LUMO轨道上的电子云除了分布在A环和萘啶环上,D环上也有明显分布.说明电子吸收光谱的最大吸收值是由分子内电荷的转移引起的,电荷主要是由A环和萘啶环向D环转移.3种化合物的ΔE值十分接近,由于化合物b的结构对称性较高,使其能量差最小ΔE=4.10 eV.而c中连接的是吸电子氯原子,阻碍了电子的跃迁,增大了轨道跃迁所需能量,使其能量差最大ΔE=4.23 eV.
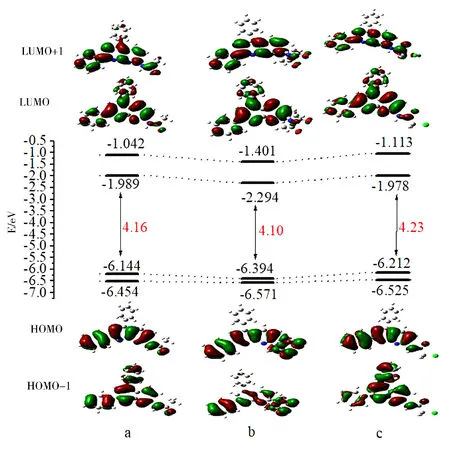
图3 3种化合物的前线分子轨道及能量
3 结 语
根据含时密度泛函理论(TD-DFT)采用B3LYP/6-31G(d)基组,首先对3种5,7-二苯基-1,8-萘啶衍生物进行结构优化得到了它们的前线分子轨道和能量,在结构优化的基础上计算了3种化合物的电子吸收光谱.计算结果和实验结果吻合,3种化合物的最大吸收值较为相近且归属为π→π*电子跃迁,它们的溶剂效应不强.与此同时证实了计算1,8-萘啶衍生物的电子吸收光谱采用B3LYP/6-31G(d)基组是合适的.
