锐钛矿型TiO2羟基化表面与H2O相互作用的分子动力学研究
2020-01-14桑丽霞陈平方
桑丽霞,雷 蕾,陈平方,王 军
(北京工业大学环境与能源工程学院,传热强化与过程节能教育部重点实验室及传热与能源利用北京市重点实验室,北京 100124)
自1972年Fujishima和Honda[1]首次利用TiO2单晶光电极实现光电催化分解水制氢以来,该半导体作为光电催化体系的光阳极材料被广泛研究. 在光电催化分解水过程中,光照射在TiO2表面使其价带中的电子向导带跃迁,由此产生的光生空穴迁移至TiO2表面发生氧化反应生成氧气,而光生电子经外电路到达光阴极发生还原反应生成氢气. 在TiO2表面进行的产氧反应是分解水反应的控制步骤[2],加快其反应速率可有效提高整体制氢效率.
TiO2在自然界主要有3种晶型——锐钛矿、金红石和板钛矿,其中锐钛矿型TiO2(anatase)在实际生产和科学研究中应用最多[3-5]. 锐钛矿型TiO2结构中丰富的氧空穴和三价钛间隙原子等缺陷可促进水分子发生解离,在表面钛、氧原子上分别形成端位羟基(terminal hydroxyl,TH)和桥羟基(bridge hydroxyl,BH),可由和频振荡光谱及原位红外光谱3 500~3 800 cm-1范围内的多个特征吸收峰证实[6-7]. 此外,X光电子能谱和傅氏转换红外光谱实验[8]得到,TiO2表面水分子受氧空穴影响易生成羟基. 羟基在半导体- 水溶液体系中的质子传递和电子转移方面起重要作用[9]. Deak等[10]利用半经验杂化泛函理论对羟基化TiO2与水分子组成的体系进行研究,结果发现提高TiO2表面桥羟基/端位羟基比例会改变半导体费米能级的位置,进而促进水分解. Kullgren等[11]通过理论研究得到半导体表面吸附OH-和H+会影响TiO2的平带电势. 并且,表面羟基还可与光生空穴结合生成羟基自由基(h++OH-→·OH),捕获光生空穴以减少载流子复合[12]. 在紫外光照射下,TiO2表面桥羟基数量增加,使得溶液等电位点降低和TiO2表面正电荷减小,而端位羟基亲水性强,具有相反作用,这2种羟基的比例会直接影响界面反应[13-14]. Predota等[15]针对离子溶液进行第一性原理分子动力学计算,对比羟基化和非羟基化TiO2表面的模拟结果发现,前者与X射线实验所得离子在界面的吸附位匹配更好. 在实验方面,Hussain等[16]由扫描隧道电子显微镜和X射线衍射等原位观察发现,液态水和金红石TiO2(110)界面第1层为(2×1)有序羟基结构(~0.38 nm),第2层为水分子. 基于实验和模拟研究可知,在表面缺陷及吸附氧和水分子的共同作用下,同时含端位羟基和桥羟基的表面结构较之前仅含桥羟基的结构更为合理,且有利于质子传递.
在锐钛矿型TiO2的晶相结构中,最稳定的是(101)面,活性最高的是(001)面[17]. 目前针对锐钛矿型TiO2(101)面与水所构建的体系研究很多. TiO2(101)表面水分子由分子吸附状态向解离状态转变需要克服一定的势垒,在表面及次表面氧空穴[18-19]等缺陷及外界热力学因素[20-21]的影响下,第1层水分子的吸附状态逐渐混乱,经过复杂的过渡反应最终在表面发生解离吸附,形成羟基化表面[22]. Geng等[23]对锐钛矿型TiO2(101)表面进行程序升温吸附研究发现,半导体表面水分子覆盖越多越容易发生解离,由此推断水分子之间的相互作用力能够促进水分解. 与锐钛矿型TiO2(101)面结构类似,(001)表面及次表面也存在缺陷可促进TiO2结构中过量电子生成和迁移,进而影响表面向羟基化转变[24-25]. Zhao等[26]利用密度泛函理论(density functional theory,DFT)和完全线性同步传输/二次同步传输方法对水分子在锐钛矿型TiO2低指数表面的吸附和解离进行研究,通过比较反应路径和中间态能量得到(001)表面水解过程为放热反应,水分子易于分解且不可逆,因此表面水分子以解离吸附为主,而(101)表面水解吸热且反应可逆,表面水分子以分子吸附为主.
本课题组已利用经典分子动力学对锐钛矿型TiO2(101)、(001)、(100)完整表面与水的相互作用进行了计算模拟[27],对比了它们的界面性质,并与晶面活性相关的耗尽层进行了关联分析. 在此基础上,本文建立锐钛矿型TiO2(101)、(001)羟基化表面,利用经典分子动力学模拟计算水分子在表面的吸附结构、运动性质以及电荷密度分布,通过与完整表面结果进行对比得到表面羟基对TiO2/H2O界面性质的影响. 羟基数量和结构与TiO2的形态[28-29]和水溶液环境(酸碱性[30]、压力[31]、温度[25]、氧气量[32]、组分[33])等众多因素有联系,通过高倍X射线光电子光谱实验和第一性原理计算结果说明,平衡状态下锐钛矿型TiO2(101)和(001)表面仅部分位置吸附有羟基[34-36],因此,本文所构建的TiO2(101)和(001)表面均为部分羟基结构.
1 计算模型的建立与可靠性验证
1.1 羟基化模型的建立
图1为锐钛矿型TiO2(101)和(001)完整表面及羟基化表面(*—OH)的结构模型. 完整TiO2(101)和(001)表面均包含未饱和配位钛原子(Ti5c)和桥氧原子(O2c),水分子在TiO2解离后生成OH-和H+分别吸附于表面Ti5c和O2c即形成表面端位羟基和桥羟基. 羟基化表面的结构参数和分子数如表1所示. Geng等[23]通过程序升温脱附实验测得TiO2(101)表面水分子分解后产生H+吸附于表面形成羟基的比例为7%,而OH-部分吸附致使表面羟基化程度高于实验结果,本文中采用10%的羟基化TiO2(101)表面. 针对(001)表面,Blomquist等[25]通过光电子分光光谱实验在较高水分子覆盖度条件下测得水分子为混合吸附状态,其中,基底表面每个氧原子对应0.4个羟基,本文所用的(001)模型表面含70个氧原子,因而对应表面羟基结构数量为28. 比较看出(001)表面羟基化比例更高,这与其表面活性高、易促进水解的性能相一致.

表1 羟基化TiO2(101)/(001)-H2O体系的结构参数和分子数
为保证体系水分子数与完整表面体系[27]相同,表面每增加一组—OH和—H吸附形成的羟基结构,溶液体系水分子数减1. 此外,在TiO2表面具体添加羟基和氢原子时需要留意其相对位置. 水分子在(101)表面分解后产生的—OH吸附于Ti5c位置,氢离子吸附于图1中的C位置[37];水分子在(001)表面分解后产生的—OH吸附于Ti5c位置,氢离子选择其斜对角O2c位置键连,非紧邻的桥氧位置[38].
整体而言,TiO2-H2O体系是3层平板模型,两边为结构相同的TiO2,中间是足够大的水分子体系,确保消除模拟过程中表面对液体体相部分的作用. 建模完成后首先对模拟体系采用最速下降法以及共轭梯度法进行几何优化得到合理结构,然后在NVT 正则系综条件下进行动力学模拟计算,温度由Nose- Hoover算法控制在298 K. 该过程时间步长为1.2 fs,总时间步数为105步,总模拟时间长度为1.2 ns. 整体结构采用周期性边界条件,优化和模拟过程均采用COMPASS(condensed-phase optimized molecular potentials for atomistic simulation studies)力场,原子间相互作用力的参数对氢原子、氧原子和钛原子均适用[39].
1.2 羟基化TiO2(101)-H2O模型可靠性验证
径向分布函数(radial distribution function,RDF)是统计力学概念,具体指其他粒子在某给定粒子周围空间的分布概率,可用于描述粒子的相关性和结构的有序性,由g(r,r′)表示. 此处的|r-r′|比较小,主要表征原子的堆积状况及各个化学键之间的距离,随着距离的增加,径向分布函数值趋向于1. 表2所示为水分子中氢、氧原子(Hw、Ow)和TiO2半导体中钛、氧原子(Ti、O)的径向分布函数第1个峰值位置.g(Ow,Ow)曲线第1个峰对应的位置为0.273 nm,文献中相应的实验值和模拟值为0.277、0.279 nm;g(Ow,Hw)曲线第1个峰对应0.097 nm,对应文献中0.100 nm;g(Ti,O)第1个峰位于距表面0.185 nm处,对应文献中的0.197、0.198 nm. 本文所得结果与文献实验、模拟结果十分接近,因此,该体系模型及所用力场能够很好地模拟半导体TiO2(101)、溶液的结构和性能.
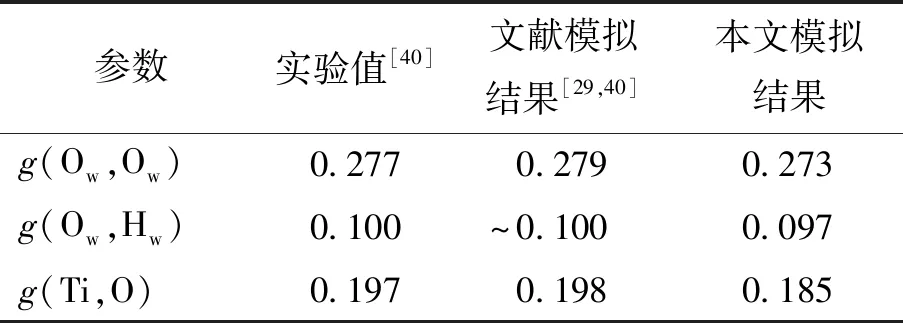
表2 羟基化TiO2(101)-H2O体系中钛、氧原子的径向分布函数第一峰值位置
图2所示为羟基中的氧原子(O*)与水分子中氢、氧原子(Hw、Ow)的径向分布函数,与文献中g(O*,Ow)和g(O*,Hw)径向分布函数曲线第1个峰的位置0.25~0.27 nm和0.1 nm符合较好[12],说明本文羟基化结构合理. 由于g(O*,Hw)函数对应的第2个峰代表与水分子相关的氢键作用,所以在0.150~0.400 nm对该曲线进行积分,所得结果为3.09,即相邻水分子之间的氢键数量为3.
图3所示为TiO2(101)表面钛、氧原子与水分子中氢、氧原子间的径向分布函数,从中看出羟基化表面对应的g(Ti5c,Ow)和g(O2c,Hw)曲线的第一峰分别出现在0.285、0.179 nm,与完整表面结果相近. 这说明水分子距半导体表面的距离受羟基影响不大. 曲线较远位置的峰代表溶液体相的水化层结构,随距离增加有序性消失.
1.3 羟基化TiO2(001)表面- 水体系模型可靠性验证
由于文献中对TiO2(101)、(001)表面的研究内容不同,本文需要对2个面进行不同的信息比较. 针对锐钛矿型TiO2(001)表面,对比了水分子及表面羟基所含氢、氧原子之间的键长,如表3所示. 文献中使用密度泛函理论进行研究所采用的体系含原子数较少[41],一般为单个羟基和少数水分子,因此所得键长仅为一个确定的数,本文采用的分子动力学研究体系很大,分子数极多,不同表面羟基表现略有差异,因而所得结果为一个范围. 具体来看,O2c-Hw和HOH-OOH键长与文献结果匹配较好,HOH-Ow比文献值稍大. 总体而言,锐钛矿型TiO2(001)羟基表面与水体系中的氢、氧原子键长合理,由此判断模型可靠.
染色色样表观深度值 (K/S),是反映染色产品表面色泽浓淡的一个指标。本实验通过在GK-02型思维士电脑测色配色仪上测定染色色样的K/S值 (测试时将待测试样叠成四层放在仪器测量部位,然后选取四个不同位置进行测试取平均值,最后记录待测试样的K/S值),来比较各染色试样的染色深度,K/S值越大,表示物体表面颜色越深。
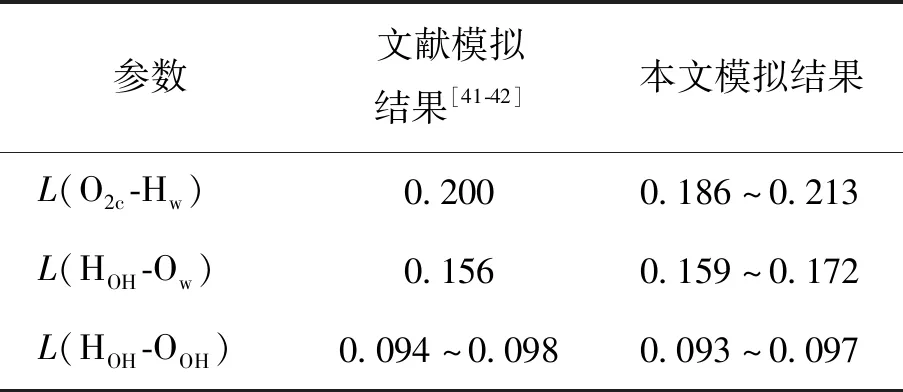
表3 羟基化TiO2(001)-H2O体系中氢、氧原子间氢键键长
图4所示为TiO2(001)表面钛、氧原子与水分子中氢、氧原子间的径向分布函数,羟基化表面对应的g(Ti5c,Ow)和g(O2c,Hw)曲线第一峰分别出现在0.380、0.200 nm,与完整表面计算结果相同,再次证明水分子距半导体表面的距离受羟基影响不大.
2 结果与讨论
2.1 羟基化TiO2(101)/(001)表面水分子的分层结构与氢键分布
氢键是与电负性较大的原子(F、Cl、O、S、N等)成键的氢原子与邻近电负性较大或带孤对电子的原子之间所产生的一种较强的非键作用,广泛存在于水溶液中. 图5是氢键形成的示意图,A、D分别代表给、受体原子. 一般认为氢键的形成条件是:给体原子与氢原子间的距离rAH小于0.25 nm,给体原子- 氢原子- 受体原子三者之间所形成的夹角θ大于90°[43]. 氢键作用可被视为氢原子及给、受体氧原子间的范德华力和静电力的综合体现,本文所采用的COMPASS力场在这方面模拟效果最好[39].
在TiO2-溶液体系中,水分子与固体表面相互作用发生单层化学吸附,然后通过氢键结合形成多层物理吸附水,最外层为相对自由的水分子束,即水在TiO2表面有3层吸附状态,分别为内层吸附的水分子或解离后吸附的羟基、中间层和最外层吸附水,其中内层吸附的水分子和羟基能够体现TiO2表面性质及其光催化活性差异[44].
图6是羟基化TiO2(101)、(001)表面水分子吸附快照图(1 200 ps),可观察到水分子大体分为3层结构,内层水分子在表面主要吸附于O2c(黄色)、桥羟基中的氢原子HBH(绿色)和端羟基中氢原子HTH(蓝色)3个位置,其中O2c位置吸附的水分子最多. 除此之外,水分子受周围环境中的水分子相互吸引力、排斥力作用存在于内层(玫红色). 中间层水分子中的氧原子和氢原子分别与第1层水分子中的氢原子、氧原子相互吸引吸附于表面(紫色),同时也存在个别水分子与表面OTH原子相互作用,受键长影响存在于外层空间(棕色). 随着水分子与半导体表面距离的增加,氢键分布逐渐稀疏,即分子间氢键减弱,相对约束减弱会造成水分子活动性增强. 上述水分子的分层结构并非严格区分,部分水分子可能同时与2类表面原子作用,或者表面羟基之间存在相互作用,该现象在其他文献中同样存在[12].
在TiO2完整表面上,水分子吸附于Ti5c位置和O2c位置,相互之间以氢键连接[27]. 表面羟基化之后,Ti5c吸附位被HBH和HTH两个吸附位取代,由此形成新的表面吸附结构.
2.2 羟基化TiO2(101)/(001)表面水分子的密度分布
分子动力学在较大时间和空间尺度下模拟半导体- 水溶液体系,与常用的密度泛函理论及第一性原理计算方法相比,能够更好地描述界面性质[37]. 图7为近表面水分子的密度分布图,据此可将近表面水溶液分为内亥姆霍兹层(IHL)、外亥姆霍兹层(OHL)和体相(Bulk). TiO2(101)羟基表面IHL、OHL及Bulk分层分别对应表面垂直距离0~0.394 nm、0.394~0.645 nm及0.645 nm之后的范围,即内、外亥姆霍兹层宽度分别为0.394和0.251 nm. TiO2(101)完整表面水分子内、外亥姆霍兹层宽度分别为0.510和0.341 nm,比较而言,表面羟基结构使得TiO2(101)表面水分子体系的内、外亥姆霍兹层宽度变窄. TiO2(001)羟基化表面对应水分子体系的内、外亥姆霍兹层层宽分别为0.510、0.451 nm,与完整表面结果(0.591、0.342 nm)相比,其内亥姆霍兹层变窄,外亥姆霍兹层变宽. 因此,TiO2(001)表面羟基作用范围更广,将外亥姆霍兹层边界由0.933 nm延伸至0.961 nm.
2.3 羟基化TiO2(101)/(001)表面水分子的运动性质
分子动力学模拟中,水分子及TiO2表面的原子在不停地变换位置,用ri(t)表示t时刻粒子i的位置. 对所有粒子位移的平方做平均得到均方根位移(mean square displacement,MSD),表示为
MSD=R(t)=〈|r(t)-r(0)|2〉
(1)
(2)
式中Di是扩散系数,用于表征水分子扩散能力.
基于上述原理,计算得到水分子在羟基化TiO2表面3个分层内的均方根位移以及扩散系数,如图8所示.
一方面,由于溶液中的水分子并非局限于特定区间,因此只能对较短时间内的均方位移进行平均以代表这一时段所处分层的扩散性质;另一方面,从统计力学角度出发,模拟时间越长、体系粒子数越多,结果越接近真实体系. 在本项研究中,选择100 ps内的水分子运动过程代表该时段水分子所处分层的扩散系数大小.D1、D2、D3分别代表内亥姆霍兹层、外亥姆霍兹层以及溶液体相区域内水分子的扩散系数. 内亥姆霍兹层内的水分子受表面约束最大,因而扩散系数最小. 外亥姆霍兹层内水分子的扩散系数有一定增加,延伸至体相,TiO2(101)和(001)羟基化表面的水分子扩散系数分别达到6.62×10-5、7.57×10-5cm2/s. 结合表面氢键分析(见图6),随表面距离增加,水分子与羟基化TiO2表面的相互作用减弱. 以TiO2/H2O界面为原点,沿界面法向方向指向水分子体系为z轴方向,平行于界面向上为y轴方向,平行于界面指向内侧为x轴方向,对整个动力学过程(1 200 ps)内TiO2表面水分子的平均速度分析,如图9所示,直观看出水分子在TiO2表面的运动表现为各向异性. 平行于TiO2表面的方向上,水分子运动速度很小且不同方向差异不大. 沿界面法线方向,近表面层的水分子速度很大,且由溶液向半导体方向运动. Zhao等[45]通过DFT模拟计算得到,水分子在TiO2(101)表面沿(010)方向的扩散能垒最小.
针对羟基化TiO2(101)-H2O体系,距界面0.104 nm处的水分子运动速度达到最大(251 m/s),而在羟基化TiO2(001)-H2O体系中,距界面0.205 nm处的水分子速度达到最大(572 m/s). 同样是羟基化表面,TiO2(001)表面水分子运动速度更大,最大速率水分子距表面更远. 结合扩散系数分析得到,水分子的运动与半导体的表面有极大关系,TiO2(001)表面结构利于水分子沿界面法向方向移动.
2.4 羟基化TiO2表面的电荷密度分布
根据表面水分子的密度分布及带电量可得电荷密度分布图,如图10所示. 首先观察TiO2(101)羟基表面结果,随着距离的增加,表面电荷首先向上达到一个极大值26.04 e/nm3,然后下降至极小值-57.03 e/nm3,反弹后达到内亥姆霍兹层第2个极大值29.74 e/nm3,同时也是电荷密度曲线的最大值. 距离延伸至外亥姆霍兹层,电荷分布波动减小,到达体相区后电荷密度收敛在零附近.
羟基化TiO2(001)近表面的电荷密度变化趋势与羟基化TiO2(101)面结果一致,首先增加到一个极大值10.14 e/nm3,然后下降至极小值-54.05 e/nm3,再回升达到内亥姆霍兹层第2个极大值23.86 e/nm3,最后在体相内收敛为零. 与完整表面相比,羟基表面所建体系中水分子的电荷密度在内亥姆霍兹层的正电荷峰值更大、负电荷密度峰值更小,说明羟基加剧了界面处的电荷扰动.
由最基本的吸附结构分析原因,TiO2表面羟基化后,Ti5c吸附位被表面羟基中的氢原子取代,近表面区域内水分子中的氧原子与之形成氢键作用,使得表面负电荷增加,表现为负电荷密度峰值减小. 为了平衡近表面区域内过剩的负电荷,正电荷密度迅速回升形成第2个较高的正电荷密度峰. 基于电荷密度分布由泊松方程估算可得表面电场及电势分布. 相比于完整表面,羟基化表面电荷极值的绝对值更大,积分后所得近表面电场更强、近表面电势差更大,利于光生电子- 空穴对的分离和传递,最终提高了光电催化分解水体系的制氢速率.
3 结论
1) TiO2(101)羟基化表面g(Ti5c,Ow)和g(O2c,Hw)曲线的第一峰分别位于距表面0.285、0.179 nm处,TiO2(001)相应的g(Ti5c,Ow)和g(O2c,Hw)曲线第一峰分别出现在0.380、0.200 nm. 与完整表面比较得到,羟基化前后表面氧、钛原子与水中氢、氧原子之间的径向分布函数结果相近. 由分子快照图直观地看出表面水分子在羟基表面形成新的吸附结构,完整表面原有Ti5c吸附位被羟基结构中的HBH和HTH取代,O2c吸附位不受表面羟基影响.
2) 水分子以氢键作用吸附于TiO2表面引起界面电荷转移,最终,水分子密度呈现内亥姆霍兹层、外亥姆霍兹层和体相3个分层结构. 对比完整表面结果,羟基使TiO2(101)表面内、外亥姆霍兹层均变窄,而TiO2(001)表面内亥姆霍兹层变窄、外亥姆霍兹层变宽. 进一步比较水分子的均方根位移及扩散系数发现,TiO2(101)表面羟基使得水分子在内亥姆霍兹层的扩散性减弱,对外层及体相影响不大,而TiO2(001)表面羟基大大增强了外亥姆霍兹层水分子的扩散性,对内层几乎无影响. 结合水分子的平均速度分布图讨论,TiO2(001)羟基表面结构利于水分子沿界面法向方向向着TiO2移动.
3) 对表面电荷密度分布进行分析,TiO2(101)和(001)羟基表面的电荷密度分布趋势一致、极值相当. 与完整表面结果相比,羟基表面所组体系中的水分子电荷密度峰的绝对值变大,以第2个正电荷密度峰增加最为明显,这说明表面羟基加剧了表面电荷扰动,进一步会增强表面电场及电势差以促进表面电子- 空穴对分离、迁移,最终表现为提升光电催化分解水体系的产氢速率.
