细胞治疗的典范:嵌合抗原受体T细胞疗法
2019-12-28钱丽玲陈蒋庆吴晓燕荆瑞瑞孙洁
钱丽玲,陈蒋庆,吴晓燕,荆瑞瑞,孙洁,2
生物工程与大健康

孙洁 博士,浙江大学医学院研究员,博士生导师。目前研究方向为CAR-T细胞杀伤、增殖及耗竭的分子机制;开发新型生物传感器与智能分子机器;优化及探索新的细胞癌症免疫疗法。成果以第一作者或通讯作者发表在、、等国际专业期刊。
细胞治疗的典范:嵌合抗原受体T细胞疗法
钱丽玲1,陈蒋庆1,吴晓燕1,荆瑞瑞1,孙洁1,2
1 浙江大学医学院附属第一医院骨髓移植中心 浙江大学医学院细胞生物学系,浙江 杭州 310058 2 浙江大学血液学研究所 浙江省干细胞与免疫治疗工程实验室,浙江 杭州 310058
嵌合抗原受体T (CAR-T) 细胞疗法是一种利用合成受体特异性靶向抗原的过继性细胞疗法(ACT),目前在血液肿瘤的治疗中有极大的临床应用价值。虽然美国食品药品监督管理局 (FDA) 已经批准两款CAR-T药物上市,但CAR-T疗法在治疗过程中仍然存在一些副作用,如细胞因子释放综合征 (CRS)、神经毒性、B细胞功能缺失等。同时,CAR-T疗法在实体瘤治疗中的效果甚微,主要原因是缺乏特异性靶点以及肿瘤微环境对CAR-T细胞功能的抑制等。文中将从CAR的结构设计、临床应用、合成生物学对新型CAR的优化来阐述应用CAR-T细胞疗法治疗肿瘤所面临的挑战及广阔前景。
细胞治疗,免疫治疗,嵌合抗原受体T细胞,合成生物学
20世纪80年代以前,药物的研发主要依赖于天然化合物和小分子药物的合成,开发了许多有效的小分子药物用于疾病治疗。随着单克隆抗体技术和蛋白质工程技术的发展,生物大分子药物如天然蛋白质或具有特定功能的重组蛋白药物应运而生,但仍然存在新的困难和挑战,由于疾病的复杂性和多样性,且存在个体差异和组织特异性,因此很难找到组织特异和个体特异的靶点。近年来,兴起了第3种治疗方法——将细胞作为药物的细胞治疗,该方法在疾病尤其是癌症的治疗方面具有划时代的意义[1]。
1 细胞治疗的分类及发展
细胞治疗是指将自体、同源异体或异种细胞经体外工程化改造和扩大培养后,输注患者体内治疗疾病的疗法。该治疗方法与传统的小分子药物和蛋白药物相比最大的特点是利用活细胞作为药物来治疗疾病,具有复杂性和可调节性等特 征[1],具体来说,将细胞作为药物具有以下优点:1) 选择性高,细胞药物能感知复杂的人体内环境,只在特定的环境中激活,以发挥相应功能,这意味着可以更大程度上限制药物的副作用;2)局部浓度高,人体代谢、药物效应动力学(Pharmacodynamics,PD) 和药物代谢动力学(Pharmacokinetics,PK) 决定了分子药物靶向性较低,它不只在病变组织或细胞内分布,而且分布于整个机体组织,这通常会造成严重的脱靶效应,而细胞药物的优势在于可主动迁移到靶组织或靶细胞内发挥作用;3) 更加个性化,由于个体差异,目前很难控制每个患者小分子药物的使用剂量,但在细胞治疗中,可应用合成生物学设计基因开关控制药物的合成或释放,也可以根据临床需要设计不同细胞药物以治疗更多疾病[1]。但细胞药物的开发和合成生物学的应用还需更多基础研究的支撑,以解决细胞药物在临床上治疗疾病种类少、副作用严重、费用昂贵等问题。
1.1 细胞治疗的分类
细胞治疗根据细胞类型可分为干细胞治疗、免疫细胞治疗和其他细胞治疗。输血是最早的细胞治疗,现已经发展到输注特定血液成分进行治疗,这使得在提高血液利用率的同时减少副作用,目前研究较多的是干细胞治疗和免疫细胞治疗。干细胞治疗是指把健康的干细胞输注到患者体内,从而修复或替换受损细胞或组织以治疗疾病的方法。目前国内外临床试验应用较广泛的是利用间充质干细胞治疗神经性疾病、糖尿病、慢性心脏疾病、肾脏病、肝脏疾病、艾滋病和癌症等疾病[2-6]。免疫细胞治疗的过程主要包括从患者或供体血液中提取免疫细胞,在体外进行工程化改造和扩增培养后,重新输注入病人体内或者直接注射到病灶处。目前用于临床试验的免疫细胞疗法主要有CAR-T、TCR-T和NK细胞疗法等[7-9]。文中主要介绍CAR-T疗法的发展、应用和前景。
1.2 CAR-T细胞治疗的发展
早在20世纪80年代,得益于过继性T细胞治疗(Adoptive T-cell therapy,ACT) 的发展,临床上已开始利用输注自体或异体供体淋巴细胞治疗一些肿瘤,如转移性黑色素瘤、复发性白血 病[10-11]等。但这种基于自然T细胞的ACT治疗存在以下不足:首先,供体T细胞可能攻击受体导致移植物抗宿主反应(Graft-versus-host disease,GVHD);同时,受体可能排斥输注的T细胞,限制它们的持久性和疗效;而且从体内分离的自然T细胞数目少,靶向特异性低,抗肿瘤效率低。这些早期ACT的临床结果表明,该疗法需要增强靶向肿瘤的特异性并提高体外扩增T细胞的数量,同时减少免疫排斥反应[12-14]。T细胞工程的出现改变了传统ACT的局限性,产生了特异性靶向肿瘤的TCR-T、CAR-T细胞[15],其中通过在T细胞表达嵌合抗原受体(Chimeric antigen receptor,CAR) 而摆脱HLA限制性的治疗方法即CAR-T疗法,该方法已经在血液瘤的治疗中取得显著的效果。本文首先简述了CAR的结构设计以及CAR-T疗法在血液瘤和实体瘤中的应用现状,然后总结了合成生物学为CAR-T细胞设计提供的新思路,最后展望CAR-T疗法在肿瘤尤其是在实体瘤治疗过程中的巨大应用潜能。
2 CAR的设计及临床应用
2.1 CAR的设计

2.2 CAR-T疗法的临床应用
临床试验已证实CAR-T细胞疗法在治疗B细胞系血液肿瘤的疗效显著,其中包括非霍奇金淋巴瘤(Non-Hodgkinlymphoma, NHL)、慢性淋巴细胞白血病(Chronic lymphocytic leukemia,CLL)、急性淋巴细胞白血病(Acute lymphoblastic leukemia,ALL),但CAR-T细胞在实体瘤治疗中仍存在一定挑战。

图1 第一、二和三代CAR的结构设计
2.2.1 CAR-T细胞在血液肿瘤中的应用
目前,CAR-T疗法在治疗ALL[22-24]中的疗效最好,应用也最广泛,其中靶向CD19的CAR在治疗成人和儿童ALL中都取得显著的疗效[25-26],文献报道,CD19 CAR-T治疗复发/难治性ALL的完全缓解率可高达90%[27]。目前FDA批准的CD19 CAR-T细胞药物分别以CD28或4-1BB为共刺激结构域[28-29]的这两种CAR-T细胞各有优点,共刺激结构域为CD28的CAR-T细胞在反应初期增殖更快,能介导更强的肿瘤杀伤能力,但同时副作用也更强,且在免疫反应后期的持久性较差;而基于4-1BB的CAR-T细胞虽然在反应初期增殖能力相比CD28 CAR较差,但持续性更好,在免疫反应后期也能维持较高细胞数量[30-32]。另外,值得注意的是,CD19 CAR-T细胞在治疗过程中也存在一些副作用,比如细胞因子释放综合征(Cytokine release syndrome,CRS),神经毒性,B细胞功能缺失,同时还存在肿瘤复发等问题[21,33-35]。已有研究表明导致CRS的主要原因是单核细胞释放的IL-1和IL-6[36],目前临床上主要采用耗竭单核细胞或使用IL-6拮抗剂(托珠单抗,Tocilizumab) 来降低CRS,也有文献研究表明改良CAR的跨膜区和铰链区结构域可减少细胞因子释放[36-40];CAR-T疗法造成的神经毒性可能是由于巨噬细胞攻击脑膜导致脑组织损伤,临床上常用IL-1抑制剂阿那白滞素(Anakinr,别名Kineret) 来控制神经毒性[36,41];由于正常B细胞表面也表达CD19,故CD19 CAR-T细胞在杀伤肿瘤细胞的同时也会攻击自身正常的B细胞,即“on-target,off-tumor”效应,造成B细胞功能缺失,目前可通过定期向患者体内输注免疫球蛋白以弥补B细胞的功能缺失[42]。尽管针对CD19抗原的CAR-T疗法疗效突出,但仍有部分患者易复发,因此需要寻找新的肿瘤靶点进行辅助治疗。CD22在大多数急性淋巴细胞白血病患者B细胞较高表达,因此目前临床上会对一些CD19 CAR-T治疗后CD19阴性的患者,开展针对CD22抗原的CAR-T治疗的临床试验,最新的结果表明在接受CD22 CAR-T细胞辅助治疗后,73%的患者获得了完全缓解(Complete remission,CR)[43]。此外,串联CD19/CD22 CAR-T目前也已用于临床试验[44],它可同时识别CD19和CD22抗原从而减少抗原逃逸引起的复发。
同时,靶向BCMA抗原的CAR-T的初步临床研究结果令人鼓舞。BCMA全称为B细胞成熟抗原(B cell maturation antigen),呈相对特异地高表达于骨髓瘤细胞表面,因此可作为多发性骨髓瘤免疫治疗的理想靶点。临床上靶向BCMA 的LCAR-B38M疗法对复发/难治性多发性骨髓瘤晚期患者的疗效较理想,总缓解率为88%,其中68%患者完全缓解(CR),但安全性上仍存在以细胞因子释放综合征(CRS) 为主的副作用[45]。
2.2.2 CAR-T疗法在治疗实体瘤中的应用
CAR-T疗法不仅在治疗血液肿瘤方面取得了突破,在治疗实体瘤方面也有一定进展。目前,在实体瘤应用方面,CAR-T疗法靶向的肿瘤抗原大多是高表达的分化抗原,例如癌胚抗原(Carcinoembryonic antigen,CEA)、前列腺特异性膜抗原(Prostate specific membrane antigen, PSMA)、双唾液酸神经节苷脂(Disialoganglioside, GD2)、糖链抗原-125 (Carbohydrate antigen-125, CA-125)、人类表皮生长因子受体2 (Human epidermal growth factor receptor-2,Her-2) 和间皮细胞抗原(Mesothelin) 等。虽然过表达的抗原种类多样,但由于其表达特异性低,CAR-T细胞对低水平表达抗原的正常细胞也高度敏感,因此治疗中存在on-target/off-tumor的副作用[21]。例如,使用高剂量的Her-2 CAR-T细胞已导致致命的副作用,其中部分原因是由于该抗原也在健康正常肺上皮和心血管细胞低表达[46]。因此,一个首选的实体肿瘤抗原靶点就要求其表达仅限于肿瘤细胞,或只发生在非常低水平的非重要正常组织。目前表皮生长因子受体Ⅲ型突变体(Epidermal growth factor receptor variant type Ⅲ,EGFRvⅢ)、糖链抗原15-3 (Carbohydrate antigen 15-3,CA 15-3)和硫酸软骨素蛋白聚糖4 (Chondroitin sulfate proteoglycan-4,CSPG-4) 分别被认为是治疗恶性胶质瘤、胰腺癌、黑色素瘤较理想的CAR靶点[21]。此外,实体瘤的肿瘤微环境(Tumor microenvironment,TME) 也会对CAR-T细胞的功能产生抑制作用。首先,肿瘤细胞的无氧糖酵解途径使其所处的TME呈现出缺氧、酸性、低营养成分的特点而不利于T细胞存活[47-49]。其次肿瘤细胞表面表达PD-L1、MHCⅡ、Gal9等配体与T细胞上的PD1、LAG-3、TIM-3等受体结合,激活T细胞上的抑制性信号通路抑制T细胞功能[50]。肿瘤微环境中的肿瘤细胞和肿瘤相关细胞如肿瘤相关成纤维细胞(Cancer-associated fibroblast,CAF)、调节性T细胞(Regulatory T cell,Treg) 等则会分泌抑制性的细胞因子,如血管内皮生长因子(Vascular endothelial growth factor,VEGF)、转化生长因子β (TGF-β),或者通过产生活性氧(ROS)、前列腺素E2 (PGE2) 和乳酸等抑制T细胞的免疫应答[51-53]。因此,CAR-T疗法在实体瘤中的应用还需要克服TME的抑制信号,增强CAR-T细胞的肿瘤识别能力、浸润能力和持久性并避免脱靶效应[21]。目前已有一些应对策略,可能会对CAR-T细胞在复杂的肿瘤微环境中发挥作用提供帮助,比如局部给药,或使用分泌IL-12的“武装CAR”、NK细胞受体CAR等[54-60]。
2.3 合成生物学方法优化CAR的设计
为了克服免疫抑制(包括阻断检查点、抑制调节性T细胞和其他髓系细胞),降低on-target/ off-tumor毒性,提高CAR-T细胞的抗肿瘤疗效,目前正在探索促进CAR-T细胞的浸润、增强CAR-T细胞的功能持久性的多种方法,其中CAR的设计与合成生物学技术的结合为设计安全 性更高、功能更强的CAR-T细胞提供了更多可能性。
2.3.1 控制CAR-T细胞的毒性和活性
目前临床上使用的CAR-T疗法所带来的器官毒性、脱靶毒性等问题亟待解决,已有报道表明严重CRS和脑水肿可引起治疗相关死亡。合成生物学的发展为更好地调控人体内的CAR-T细胞提供了新的思路。其中一种策略是通过小分子药物来控制CAR-T细胞自凋亡或抗原抗体结合来降低CAR-T细胞治疗过程中的毒性反应。其具体设计有以下几种方法:其一,在CAR-T细胞内加入相应的信号蛋白作为分子开关,实现对T细胞可逆调控[61]或“自杀”基因如单纯疱疹病毒胸苷激酶(HSV-TK) 基因,一旦CAR-T细胞在患者体内发生不良反应,通过施加药物介导T细胞的自杀基因激活诱导CAR-T细胞凋亡[62-63]。其二,利用小分子药物,如AP1903二聚化诱导型Caspase 9 (iCasp9) 激活T细胞的自杀开关,诱导其发生凋亡终止CAR-T细胞发挥作用。其三,某些小分子药物能够通过介导肿瘤抗原和CAR之间的结合来调节CAR-T细胞识别肿瘤抗原的能力。最后,将CAR的scFv与信号转导结构域分隔开,两者之间依靠小分子二聚化的可逆结合得以更灵活地调控CAR-T细胞的功能[64-65]。
2.3.2 增强CAR-T细胞肿瘤识别的特异性
利用合成生物学制备多靶点CAR或Boolean logic-gated CAR,用于识别一种或两种肿瘤抗原的方式来增强工程化的CAR-T细胞识别肿瘤抗原的能力,比如合成Notch (synNotch) 受体、嵌合共刺激受体(CCR)、抑制性嵌合抗原受体(iCAR) 等。其中SynNotch受体是一种精确识别肿瘤抗原的设计,该策略利用Notch受体独特的信号传导机制,即抗原A特异性的synNotch在结合了肿瘤抗原A之后,转录因子被切割激活,进而转运到细胞核引起了抗原B特异性的CAR的表达,最终与肿瘤表面的抗原B结合,T细胞被激活[66-68](图2A)。此外,synNotch受体系统还具有整合多个细胞外信号的潜能[69]。CCR是将用于T细胞活化的一代CAR和用于共刺激的CCR组合到一起,只有当两种抗原同时存在于肿瘤细胞表面时,CAR-T细胞才能激活并介导肿瘤杀伤(图2B)。这样的设计增强了CAR-T细胞识别肿瘤细胞抗原的特异性,减轻副作用[70]。SynNotch受体和CCR的识别逻辑都是“AND gate”。另一种策略则是将识别不同肿瘤抗原的scFv串联在一起,只要肿瘤细胞表达其中一个抗原[71],就能活化T细胞介导肿瘤杀伤(图2C),即“OR gate”。基于CTLA-4和PD-1的iCAR通过识别正常细胞表面的抗原来发挥作用[72],是一种“AND OR NOT gate”,CAR-T细胞一旦识别在正常细胞表面的抗原,就会传递抑制信号抑制T细胞的活化,因此可有效预防CAR-T细胞对正常细胞的不良反应(图2D)。SUPRA (Split,universal and programmable) CARs则整合了以上3种肿瘤识别逻辑[71,73-75],这是一种通用型的CAR,该策略是将CAR分成两个部分Zip CAR (不含scFv) 和Zip Fv (不含信号转导结构域),二者能通过匹配的亮氨酸拉链结合以形成完整的CAR,且可通过使用不同组合的抗原靶向部分控制对肿瘤抗原的识别能力[74],SUPRA CAR的开发表明多个高级逻辑识别抗原功能可在单个集成系统中实现(图2E)。
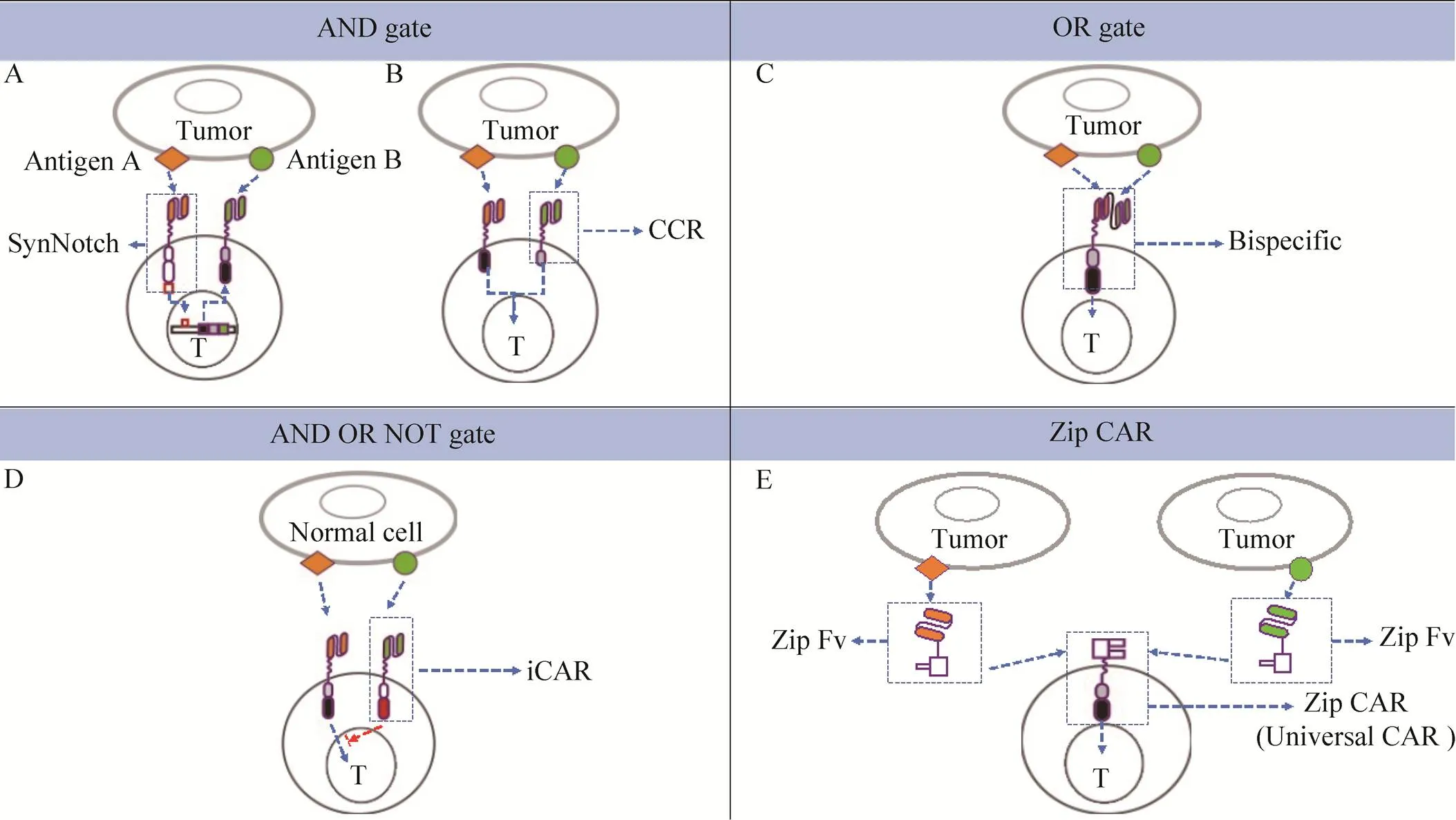
图2 运用合成生物学设计的新型CARs
目前,对这些CAR的研究还处在初级阶段,在向临床研究转化的过程中,还存在许多困难和挑战等待研究者去克服。
3 CAR-T治疗的前景
肿瘤免疫治疗于2013年被杂志评为十大科技突破之首,其中CAR-T作为具有抗原靶向性且有一定持久性的“活细胞药物”,可重编程T 细胞的效应和分化等功能,在接触抗原后增殖并发挥抗肿瘤作用,尤其是CAR-T疗法在B细胞血液瘤中取得了突破,给复发/难治性白血病及淋巴瘤患者带来了希望[26-27]。但肿瘤微环境的免疫抑制[76]、抗原表达缺失或下调以及CAR-T细胞持久性不足等导致仍有患者复发。此外,初步临床结果表明CAR-T疗法在对标准治疗有耐药性的多发性骨髓瘤治疗中取得重大进展[45]。虽然CAR-T在实体瘤治疗上仍缺乏重大突破,但综合目前CAR-T治疗在临床以及以下各个相关领域的进展,不可否认的是CAR-T疗法具有广阔前景:1) 随着基因编辑技术和合成生物学的发展,CAR结构的设计更具灵活性和多元化。2) 已有研究表明敲除T细胞受体的同时将CAR插入T细胞受体位点可延迟T细胞耗竭从而增强抗癌作用,这为未来CAR-T设计提供重要思路[44]。3) 选择最优的T细胞亚群,如调整CD4/CD8 T细胞比率和初始T细胞/效应T细胞比率用于CAR治疗,将进一步提高CAR治疗的疗效和安全性[21]。4) 此外,初步临床结果表明CAR-T治疗与PD-1/PD-L1抑制剂联用,或两个不同靶点的CAR-T联用在淋巴瘤治疗上有较好成效[21,77-78]。5) 为了使CAR-T细胞治疗成为更普遍的治疗手段,已经出现了“通用型”CAR-T细胞,并在机体表现出很强免疫抑制的B-ALL患儿中取得了成功[79]。开发出具有更优性能的“通用型”CAR-T细胞,重点是避免机体和T细胞的相互排斥,因此通过使用基因编辑技术沉默基因、Ⅰ类基因的同种异体T细胞,不仅能有效消除可能造成的GVHD[76],经过改造后,在患者急需治疗时能提供大量可用的现成CAR-T细胞,这对临床治疗具有重要意义。6) 对肿瘤发病、复发机制以及对T细胞和CAR-T细胞功能机制的知识体系的扩展和更新也为CAR-T治疗提供深厚的理论基础。随着CAR-T基础研究与临床实践的不断深入,CAR-T细胞必将在肿瘤生物细胞免疫治疗中发挥越来越重要的作用。
[1] Fischbach MA, Bluestone JA, Lim WA. Cell-based therapeutics: the next pillar of medicine. Sci Transl Med, 2013, 5(179): 179ps7.
[2] Couri CEB, Voltarelli JC. Stem cell therapy for type 1 diabetes mellitus: a review of recent clinical trials. Diabetol Metab Syndr, 2009, 1: 19.
[3] Eom YW, Shim KY, Baik SK. Mesenchymal stem cell therapy for liver fibrosis. Korean J Intern Med, 2015, 30(5): 580–589.
[4] Nguyen PK, Rhee JW, Wu JC. Adult stem cell therapy and heart failure, 2000 to 2016: a systematic review. JAMA Cardiol, 2016, 1(7): 831–841.
[5] Shroff G, Dhanda Titus J, Shroff R. A review of the emerging potential therapy for neurological disorders: human embryonic stem cell therapy. Am J Stem Cells, 2017, 6(1): 1–12.
[6] Zhang Z, Fu JL, Xu XS, et al. Safety and immunological responses to human mesenchymal stem cell therapy in difficult-to-treat HIV-1-infected patients. AIDS, 2013, 27(8): 1283–1293.
[7] Ikeda H. T-cell adoptive immunotherapy using tumor-infiltrating T cells and genetically engineered TCR-T cells. International Immunol, 2016, 28(7): 349–353.
[8] Rezvani K, Rouce R, Liu EL, et al. Engineering natural killer cells for cancer immunotherapy. Mol Ther, 2017, 25(8): 1769–1781.
[9] Roncarolo MG, Battaglia M. Regulatory T-cell immunotherapy for tolerance to self antigens and alloantigens in humans. Nat Rev Immunol, 2007, 7(8): 585–598.
[10] Mttchison NA, Dube OL. Studies on the immunological response to foreign tumor transplants in the mouse. II. The relation between hemagglutinating antibody and graft resistance in the normal mouse and mice pretreated with tissue preparations. J Exp Med, 1955, 102(2): 179–197.
[11] Rosenberg SA, Packard BS, Aebersold PM, et al. Use of tumor-infiltrating lymphocytes and interleukin-2 in the immunotherapy of patients with metastatic melanoma. A preliminary report. New Engl J Med, 1988, 319(25): 1676–1680.
[12] Greenberg PD. Adoptive T cell therapy of tumors: mechanisms operative in the recognition and elimination of tumor cells. Adv Immunol, 1991, 49: 281–355.
[13] Melief CJM. Tumor eradication by adoptive transfer of cytototic T lymphocytes. Adv Cancer Res, 1992, 58: 143–175.
[14] Old LJ. Tumor immunology: the first century. Curr Opin Immunol, 1992, 4(5): 603–607.
[15] Sadelain M, Rivière I, Riddell S. Therapeutic T cell engineering. Nature, 2017, 545(7655): 423–431.
[16] Thistlethwaite FC, Gilham DE, Guest RD, et al. The clinical efficacy of first-generation carcinoembryonic antigen (CEACAM5)-specific CAR T cells is limited by poor persistence and transient pre-conditioning-dependent respiratory toxicity. Cancer Immunol Immunother, 2017, 66(11): 1425–1436.
[17] Ghobadi A. Chimeric antigen receptor T cell therapy for non-Hodgkin lymphoma. Curr Res Transl Med, 2018, 66(2): 43–49.
[18] Carpenito C, Milone MC, Hassan R, et al. Control of large, established tumor xenografts with genetically retargeted human T cells containing CD28 and CD137 domains. Proc Natl Acad Sci USA, 2009, 106(9): 3360–3365.
[19] Wang JJ, Jensen M, Lin YK, et al. Optimizing adoptive polyclonal T cell immunotherapy of lymphomas, using a chimeric T cell receptor possessing CD28 and CD137 costimulatory domains. Hum Gene Ther, 2007, 18(8): 712–725.
[20] Pulè MA, Straathof KC, Dotti G, et al. A chimeric T cell antigen receptor that augments cytokine release and supports clonal expansion of primary human T cells. Mol Ther, 2005, 12(5): 933–941.
[21] Sadelain M. Chimeric antigen receptors: a paradigm shift in immunotherapy. Annu Rev Canc Biol, 2017, 1: 447–466.
[22] Brentjens RJ, Davila ML, Riviere I, et al. CD19-targeted T cells rapidly induce molecular remissions in adults with chemotherapy-refractory acute lymphoblastic leukemia. Sci Transl Med, 2013, 5(177): 177ra38.
[23] Davila ML, Riviere I, Wang XY, et al. Efficacy and toxicity management of 19–28z CAR T cell therapy in B cell acute lymphoblastic leukemia. Sci Transl Med, 2014, 6(224): 224ra25.
[24] Lee DW, Kochenderfer JN, Stetler-Stevenson M, et al. T cells expressing CD19 chimeric antigen receptors for acute lymphoblastic leukaemia in children and young adults: a phase 1 dose-escalation trial. Lancet, 2015, 385(9967): 517–528.
[25] Gardner RA, Finney O, Annesley C, et al. Intent-to-treat leukemia remission by CD19 CAR T cells of defined formulation and dose in children and young adults. Blood, 2017, 129(25): 3322–3331.
[26] Sadelain M. CAR therapy: the CD19 paradigm. J Clin Invest, 2015, 125(9): 3392–3400.
[27] Maude SL, Frey N, Shaw PA, et al. Chimeric antigen receptor T cells for sustained remissions in leukemia. New Engl J Med, 2014, 371(16): 1507–1517.
[28] Forsberg MH, Das A, Saha K, et al. The potential of CAR T therapy for relapsed or refractory pediatric and young adult B-cell ALL. Ther Clin Risk Manag, 2018, 14: 1573–1584.
[29] Hombach AA, Holzinger A, Abken H. The weal and woe of costimulation in the adoptive therapy of cancer with chimeric antigen receptor (CAR)-redirected T cells. Curr Mol Med, 2013, 13(7): 1079–1088.
[30] Salter AI, Ivey RG, Kennedy JJ, et al. Phosphoproteomic analysis of chimeric antigen receptor signaling reveals kinetic and quantitative differences that affect cell function. Sci Signal, 2018, 11(544): eaat6753.
[31] Maloney DG. Anti-CD19 CAR T cell therapy for lymphoma-off to the races!. Nat Rev Clin Oncol, 2019, 16(5): 279–280.
[32] Kawalekar OU, Posey AD Jr, Fraietta J, et al. Distinct signaling by chimeric antigen receptors (CARs) containing CD28 signaling domain versus 4–1BB in primary human T cells. Blood, 2013, 122(21): 2902.
[33] Bonifant CL, Jackson HJ, Brentjens RJ, et al. Toxicity and management in CAR T-cell therapy. Mol Ther-Oncolytics, 2016, 3: 16011.
[34] Porter DL, Hwang WT, Frey NV, et al. Chimeric antigen receptor T cells persist and induce sustained remissions in relapsed refractory chronic lymphocytic leukemia. Sci Transl Med, 2015, 7(303): 303ra139.
[35] Maude SL, Barrett D, Teachey DT, et al. Managing cytokine release syndrome associated with novel T cell-engaging therapies. Cancer J, 2014, 20(2): 119–122.
[36] Norelli M, Camisa B, Barbiera G, et al. Monocyte-derived IL-1 and IL-6 are differentially required for cytokine-release syndrome and neurotoxicity due to CAR T cells. Nat Med, 2018, 24(6): 739–748.
[37] Maude SL, Laetsch TW, Buechner J, et al. Tisagenlecleucel in children and young adults with B-cell lymphoblastic leukemia. New Engl J Med, 2018, 378(5): 439–448.
[38] Turtle CJ, Hanafi LA, Berger C, et al. CD19 CAR-T cells of defined CD4+: CD8+composition in adult B cell ALL patients. J Clin Invest, 2016, 126(6): 2123–2138.
[39] Park JH, Riviere I, Gonen M, et al. Long-term follow-up of CD19 CAR therapy in acute lymphoblastic leukemia. New Engl J Med, 2018, 378(5): 449–459.
[40] Ying ZT, Huang XF, Xiang XY, et al. A safe and potent anti-CD19 CAR T cell therapy. Nat Med, 2019, 25(6): 947–953.
[41] Fox E, Jayaprakash N, Pham TH, et al. The serum and cerebrospinal fluid pharmacokinetics of anakinra after intravenous administration to non-human primates. J Neuroimmunol, 2010, 223(1/2): 138–140.
[42] Tasian SK, Gardner RA. CD19-redirected chimeric antigen receptor-modified T cells: a promising immunotherapy for children and adults with B-cell acute lymphoblastic leukemia (ALL). Ther Adv Hematol, 2015, 6(5): 228–241.
[43] Fry TJ, Shah NN, Orentas RJ, et al. CD22-targeted CAR T cells induce remission in B-ALL that is naive or resistant to CD19-targeted CAR immunotherapy. Nat Med, 2018, 24(1): 20–28.
[44] Schneider D, Xiong Y, Wu DR, et al. A tandem CD19/CD20 CAR lentiviral vector drives on-target and off-target antigen modulation in leukemia cell lines. J Immunother Cancer, 2017, 5: 42.
[45] Zhao WH, Liu J, Wang BY, et al. A phase 1, open-label study of LCAR-B38M, a chimeric antigen receptor T cell therapy directed against B cell maturation antigen, in patients with relapsed or refractory multiple myeloma. J Hematol Oncol, 2018, 11: 141.
[46] Morgan RA, Yang JC, Kitano M, et al. Case report of a serious adverse event following the administration of T cells transduced with a chimeric antigen receptor recognizing ERBB2. Mol Ther, 2010, 18(4): 843–851.
[47] Hirata E, Sahai E. Tumor Microenvironment and Differential Responses to Therapy. Cold Spring Harb Perspect Med, 2017, 7(7): a026781.
[48] Newick K, O’brien S, Moon E, et al. CAR T cell therapy for solid tumors. Annu Rev Med, 2017, 68: 139–152.
[49] Renner K, Singer K, Koehl GE, et al. Metabolic hallmarks of tumor and immune cells in the tumor microenvironment. Front Immunol, 2017, 8: 248.
[50] Wherry EJ, Kurachi M. Molecular and cellular insights into T cell exhaustion. Nat Rev Immunol, 2015, 15(8): 486–499.
[51] Martinez M, Moon EK. CAR T cells for solid tumors: new strategies for finding, infiltrating, and surviving in the tumor microenvironment. Front Immunol, 2019, 10: 128.
[52] Morrot A, Da Fonseca LM, Salustiano EJ, et al. Metabolic symbiosis and immunomodulation: how tumor cell-derived lactate may disturb innate and adaptive immune responses. Front Oncol, 2018, 8: 81.
[53] Ohta A. A metabolic immune checkpoint: adenosine in tumor microenvironment. Front Immunol, 2016, 7: 109.
[54] Mardiana S, Solomon BJ, Darcy PK, et al. Supercharging adoptive T cell therapy to overcome solid tumor-induced immunosuppression. Sci Transl Med, 2019, 11(495): eaaw2293.
[55] Hinrichs CS, Restifo NP. Reassessing target antigens for adoptive T-cell therapy. Nat Biotechnol, 2013, 31(11): 999–1008.
[56] Klebanoff CA, Rosenberg SA, Restifo NP. Prospects for gene-engineered T cell immunotherapy for solid cancers. Nat Med, 2016, 22(1): 26–36.
[57] Morello A, Sadelain M, Adusumilli PS. Mesothelin-targeted CARs: driving T cells to solid tumors. Cancer Discov, 2016, 6(2): 133–146.
[58] Moon EK, Carpenito C, Sun J, et al. Expression of a functional CCR2 receptor enhances tumor localization and tumor eradication by retargeted human T cells expressing a mesothelin-specific chimeric antibody receptor. Clin Cancer Res, 2011, 17(14): 4719–4730.
[59] Chmielewski M, Kopecky C, Hombach AA, et al. IL-12 release by engineered T cells expressing chimeric antigen receptors can effectively muster an antigen-independent macrophage response on tumor cells that have shut down tumor antigen expression. Cancer Res, 2011, 71(17): 5697–5706.
[60] Spear P, Barber A, Rynda-Apple A, et al. NKG2D CAR T-cell therapy inhibits the growth of NKG2D ligand heterogeneous tumors. Immunol Cell Biol, 2013, 91(6): 435–440.
[61] Wei P, Wong WW, Park JS, et al. Bacterial virulence proteins as tools to rewire kinase pathways in yeast and immune cells. Nature, 2012, 488(7411): 384–388.
[62] Bonini C, Ferrari G, Verzeletti S, et al. HSV-TK gene transfer into donor lymphocytes for control of allogeneic graft-versus-leukemia. Science, 1997, 276(5319): 1719–1724.
[63] Tiberghien P, Reynolds CW, Keller J, et al. Ganciclovir treatment of herpes simplex thymidine kinase-transduced primary T lymphocytes: an approach for specificdonor T-cell depletion after bone marrow transplantation? Blood, 1994, 84(4): 1333–1341.
[64] Sun J, Sadelain M. The quest for spatio-temporal control of CAR T cells. Cell Res, 2015, 25(12): 1281–1282.
[65] Wu CY, Roybal KT, Puchner EM, et al. Remote control of therapeutic T cells through a small molecule-gated chimeric receptor. Science, 2015, 350(6258): aab4077.
[66] Gordon WR, Zimmerman B, He L, et al. Mechanical allostery: evidence for a force requirement in the proteolytic activation of notch. Dev Cell, 2015, 33(6): 729–736.
[67] Morsut L, Roybal KT, Xiong X, et al. Engineering customized cell sensing and response behaviors using synthetic notch receptors. Cell, 2016, 164(4): 780–791.
[68] Roybal KT, Rupp LJ, Morsut L, et al. Precision tumor recognition by T cells with combinatorial antigen-sensing circuits. Cell, 2016, 164(4): 770–779.
[69] Themeli M, Sadelain M. Combinatorial antigen targeting: ideal T-cell sensing and anti-tumor response. Trends Mol Med, 2016, 22(4): 271–273.
[70] Kloss CC, Condomines M, Cartellieri M, et al. Combinatorial antigen recognition with balanced signaling promotes selective tumor eradication by engineered T cells. Nat Biotechnol, 2013, 31(1): 71–75.
[71] Wu MR, Jusiak B, Lu TK. Engineering advanced cancer therapies with synthetic biology. Nat Rev Cancer, 2019, 19(4): 187–195.
[72] Fedorov VD, Themeli M, Sadelain M. PD-1- and CTLA-4-based inhibitory chimeric antigen receptors (iCARs) divert off-target immunotherapy responses. Sci Transl Med, 2013, 5(215): 215ra172.
[73] Chen YY. Increasing T cell versatility with SUPRA CARs. Cell, 2018, 173(6): 1316–1317.
[74] Cho JH, Collins JJ, Wong WW. Universal chimeric antigen receptors for multiplexed and logical control of T cell responses. Cell, 2018, 173(6): 1426–1438.
[75] Chopane A, Gupta S, Ajit A, et al. Design and analysis of plastic gears in rack and pinion steering system for formula supra car. Mater Today-Proc, 2018, 5(2): 5154–5164.
[76] Scarfò I, Maus MV. Current approaches to increase CAR T cell potency in solid tumors: targeting the tumor microenvironment. J Immunother Cancer, 2017, 5(1): 28.
[77] Ribas A, Wolchok JD. Cancer immunotherapy using checkpoint blockade. Science, 2018, 359(6382): 1350–1355.
[78] Otahál P, Průková D, Král V, et al. Lenalidomide enhances antitumor functions of chimeric antigen receptor modified T cells. Oncoimmunology, 2016, 5(4): e1115940.
[79] June CH, Sadelain M. Chimeric antigen receptor therapy. New Engl J Med, 2018, 379(1): 64–73.
Cell therapy’s poster child: Chimeric antigen receptor T cell therapy
Liling Qian1, Jiangqing Chen1, Xiaoyan Wu1, Ruirui Jing1, and Jie Sun1,2
1 Bone Marrow Transplantation Center of the First Affiliated Hospital, and Department of Cell Biology, Zhejiang University School of Medicine, Hangzhou 310058, Zhejiang, China 2 Institute of Hematology, Zhejiang University& Laboratory of Stem Cell and Immunotherapy Engineering, Hangzhou 310058, Zhejiang, China
Chimeric antigen receptor T (CAR-T) cell therapy, which adoptively transfers engineered T cells expressing synthetic receptors to target specific antigens, has achieved great clinical success in treating hematological malignancies. Though FDA has approved two CAR-T products, CAR-T therapy can cause some side effects, such as cytokine release syndrome (CRS), neurotoxicity and B cell aplasia. Meanwhile, lacking tumor specific antigen and the suppressive tumor environment limit the efficacy of CAR-T therapy in solid tumor. This review focuses on the structural components, clinical applications and synthetic biology approaches on CAR-T cell design, and summarizes the challenges and perspectives of CAR-T therapy as a revolutionary cancer immunotherapy.
cell therapy, immunotherapy, chimeric antigen receptor T cell, synthetic biology
July 1, 2019;
September 4, 2019
s:Jie Sun. Tel: +86-571-88208509; Fax: +86-571-88208094; E-mail: sunj4@zju.edu.cn
2019-10-10
http://kns.cnki.net/kcms/detail/11.1998.Q.20191010.0940.003.html
钱丽玲, 陈蒋庆, 吴晓燕, 等. 细胞治疗的典范:嵌合抗原受体T细胞疗法. 生物工程学报, 2019, 35(12): 2339–2349.
Qian LL, Chen JQ, Wu XY, et al. Cell therapy’s poster child: Chimeric antigen receptor T cell therapy. Chin J Biotech, 2019, 35(12): 2339–2349.
(本文责编 陈宏宇)
