新生儿葡萄糖/半乳糖吸收不良症2例病例报告并文献复习
2019-12-19彭小敏蒋思远程国强王慧君吴冰冰周文浩
彭小敏 张 澜 蒋思远 曹 云 程国强 王慧君 吴冰冰,4 杨 琳,5 周文浩,3
1 病例资料
例1:男,29 d,因“生后纳差、体重减轻至今”入复旦大学附属儿科医院(我院)。患儿系G2P2,胎龄38+6周,剖宫产娩出,出生体重3 150 g,Apgar评分不详,否认窒息抢救史。出生后母乳喂养,纳奶差,每日解稀水样大便10~12次,未见脓液和血丝,入院前2 d添加配方奶,每次10~20 mL,2~2.5 h进食1次,大便性状无改善,尿量减少。
家族史:患儿爸爸及爷爷进食奶制品后会出现腹泻。G1P1为女孩,3岁,体健。
体格检查:神志清楚,足月儿貌,体重2 320 g,反应可,皮下脂肪菲薄,全身皮肤无黄染,无湿疹,无水肿,臀部皮肤有破损,浅表淋巴结未及肿大。头颅圆,前囟平软(1.5 cm×1.5 cm),双侧瞳孔等大等圆(3 mm×3 mm),对光反应灵敏。心肺听诊未见异常,腹部平软,肝脾未及肿大,肠鸣音正常。四肢活动可,肌力及肌张力正常。拥抱反射、吸吮反射和握持反射可引出。
实验室检查:入院血气分析pH 7.232,PO295.1 mmHg,PCO228.2 mmHg,乳酸0.9 mmol·L-1,BE -15.7 mmol·L-1,Na+183.0 mmol·L-1,K+2.9 mmol·L-1, Cl-148.0 mmol·L-1, HCO3-13.3 mmol·L-1。单纯疱疹病毒Ⅰ型和Ⅱ型、巨细胞病毒、EB病毒、轮状病毒、肠道腺病毒和真菌葡聚糖阴性。血常规、CRP、血培养、尿培养和多次粪培养均未见异常。甲状腺功能、血氨、血尿串联质谱和肾上腺激素水平均正常。免疫功能、CD系列和免疫球蛋白水平正常。
入院后予禁食,纠正脱水和电解质紊乱,先后予深度水解蛋白无乳糖配方奶(蔼儿舒)、氨基酸特殊配方奶(纽康特)和乳蛋白深度水解无乳糖配方奶(纽太特)喂养,大便次数随着奶量增长而增加,每天解黄绿色稀水样便6~10次,有酸臭味,无便血,Na+有上升趋势,体重无增长。果糖激发试验阳性,葡萄糖激发试验阴性,但出现体温波动,热峰38.5℃,考虑与葡萄糖吸收障碍有关。入院第19 d肾脏B超提示双肾结构欠清,椎体内结晶可能。入院第32 d予果糖+脂肪乳剂喂养,大便稀糊状,第37 d予含果糖特殊配方奶Galactomin 19喂养,并添加维生素AD和肉碱,腹泻渐缓解,体重缓慢增长,至第42 d出院时体重3 300 g,每天解3~5次黄色糊状便。1月后门诊随访患儿视听反应可,会吃手,抬头可。
例2:女,7 d,因“腹泻5 d”入我院。患儿系G3P2,胎龄39+1周,剖宫产娩出,出生体重3 080 g,Apgar评分9~10分,否认窒息抢救史。生后即开奶,母乳喂养不足添加雀巢部分水解配方奶喂养,纳奶可,无呕吐。生后第3 d开始出现腹泻,每日解15~20次金黄色稀糊样或水样便,量不等,无黏胨及脓血,小便量稍少(具体量未计)。当地医院予纽太特喂养,同时予纠酸和补液支持等治疗,大便次数仍多,较稀。
家族史:父母体健,否认近亲结婚。哥哥8岁,生后4~5 d开始出现腹泻,幼儿时反复腹泻,添加辅食后有所好转,现智力正常。
体格检查:足月儿貌,反应可,全身皮肤红润,无湿疹,无脓疱疹,无水肿,浅表淋巴结未及肿大。头颅圆,前囟平软(1.5 cm×1.5 cm),颅缝平软,双侧瞳孔等大等圆(3 mm×3 mm),对光反应灵敏。心肺听诊未见异常,腹部平软,肝脾未及肿大,肠鸣音正常。四肢活动可,肌力及肌张力正常。拥抱反射、吸吮反射和握持反射正常。
实验室检查:入我院时血气分析pH 7.545,PO294 mmHg,PCO219.3 mmHg,乳酸1.8 mmol·L-1,BE -5.8 mmol·L-1,K+4.4 mmol·L-1,Na+141.0 mmol·L-1,Cl-113.0 mmol·L-1。轮状病毒和肠道腺病毒阴性。粪常规:乳糖阴性,隐血阴性,少量脂肪球。免疫功能、CD系列和免疫球蛋白水平正常。甲状腺功能和血氨均正常。
入院后予蔼儿舒奶喂养,酪酸梭菌二连活菌调节肠道菌群,蒙脱石散保护胃肠道黏膜,喂养耐受,每日解5~8次稀糊状便。3月后门诊随访时家属诉患儿每日解稀水样便10次,视听反应可,能独坐。
2 全外显子组检测
经患儿父母知情同意,行家系全外显子组测序(WES)。采集患儿及其父母外周静脉血各2 mL(例2同时采集患儿哥哥血样),抽提基因组DNA(德国Qiagen公司miniblood全血试剂盒)。利用Agilent SureSelct Human All Exon V5试剂盒进行外显子捕获、建库,Illumina HiSeq2000平台对全基因组编码区外显子进行测序,捕获目标序列50 Mb,总体测序覆盖度达95%。采用我院转化医学中心自主建立的数据分析流程[1],结合WuXiNextCODE分析软件进行数据分析。通过Burrows-Wheeler Aligner (BWA)将测序数据与NCBI RefSeq (GRCh37/hg19)进行匹配比对,利用ANNOVAR、VEP和WuXiNextCODE软件注释变异数据,包括NCBI RefSeq和SwissPort进行基因注释,HGMD、OMIM和ClinVar进行疾病相关注释,千人基因组计划、gnomAD、ExAC和内部数据库进行突变频率注释,SIFT、PolyPhen-2和MutationTaster软件进行突变预测等。通过变异频率和变异类别的筛选以及与疾病的关系,筛选出可能的致病变异。
结果显示,2例患儿均检测到SLC5A1基因(NM_000343.3)的复合杂合变异(图1)。例1携带c.325dupG(p.V109GfsTer14)和c.1107_1109delAGT(p.V371del)变异,前者来自父亲,为移码变异,导致氨基酸编码提前终止,公共数据库和内部数据库均未见报道;后者来自母亲,是HGMD数据库已收录的致病变异。例2及其哥哥均携带c.781G>A(p.D261N)和c.1298T>C(p.L433P)变异,前者来自父亲,ExAc数据库有2条杂合记录,gnomAD有6条杂合记录,SIFT和MutationTaster预测为有害变异,PolyPhen-2预测为很可能有害;后者来自母亲,公共数据库和内部数据库未见报道,软件均预测为有害变异(表1)。
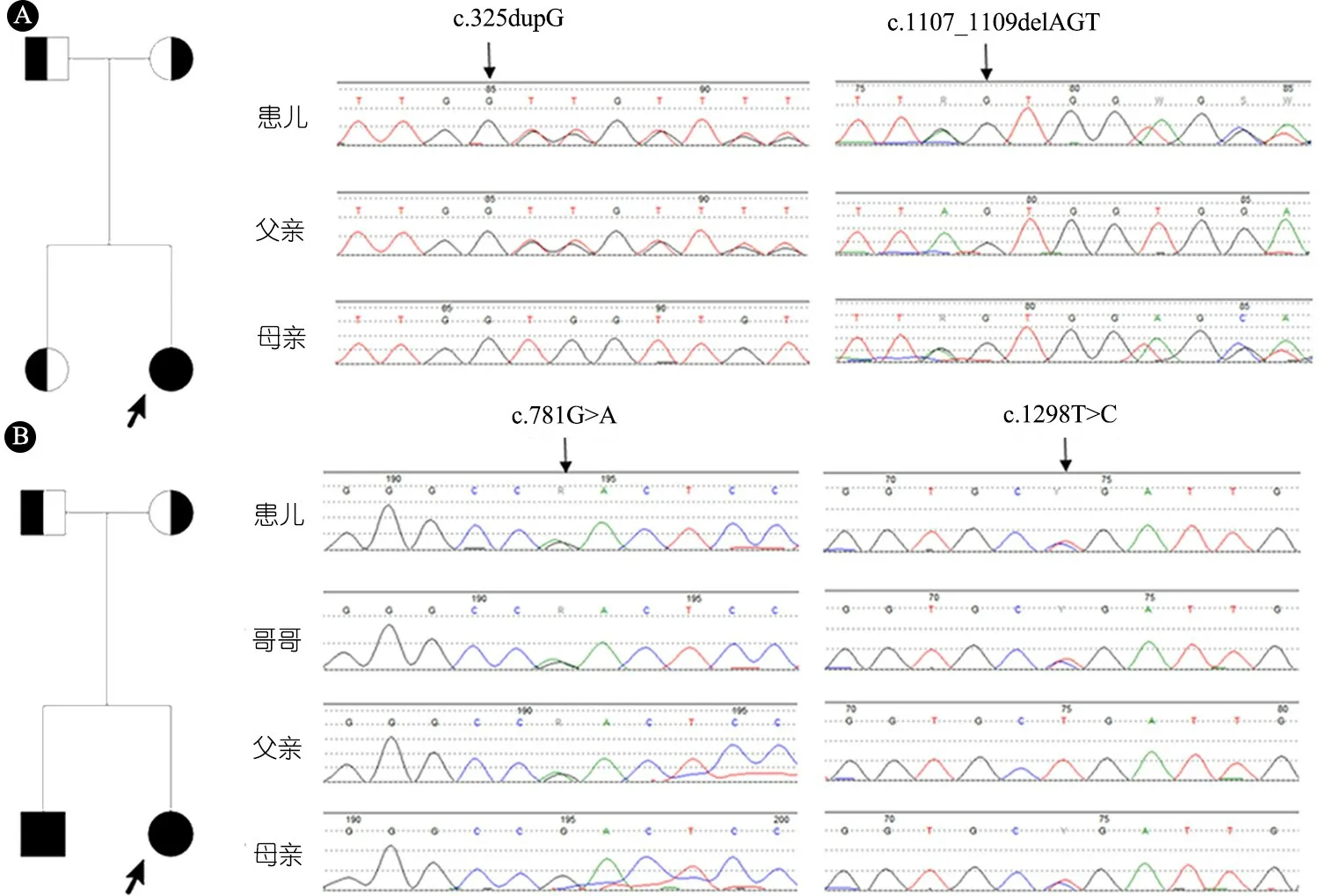
图1SLC5A1基因变异患儿家系图及Sanger测序结果
注 A:例1家系图及Sanger验证结果;B:例2家系图及Sanger验证结果
注 Het Hom:杂合样本数|纯合样本数
3 文献复习
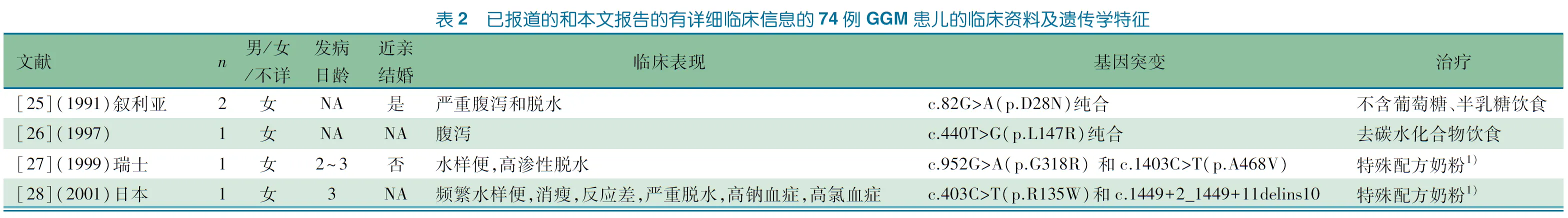
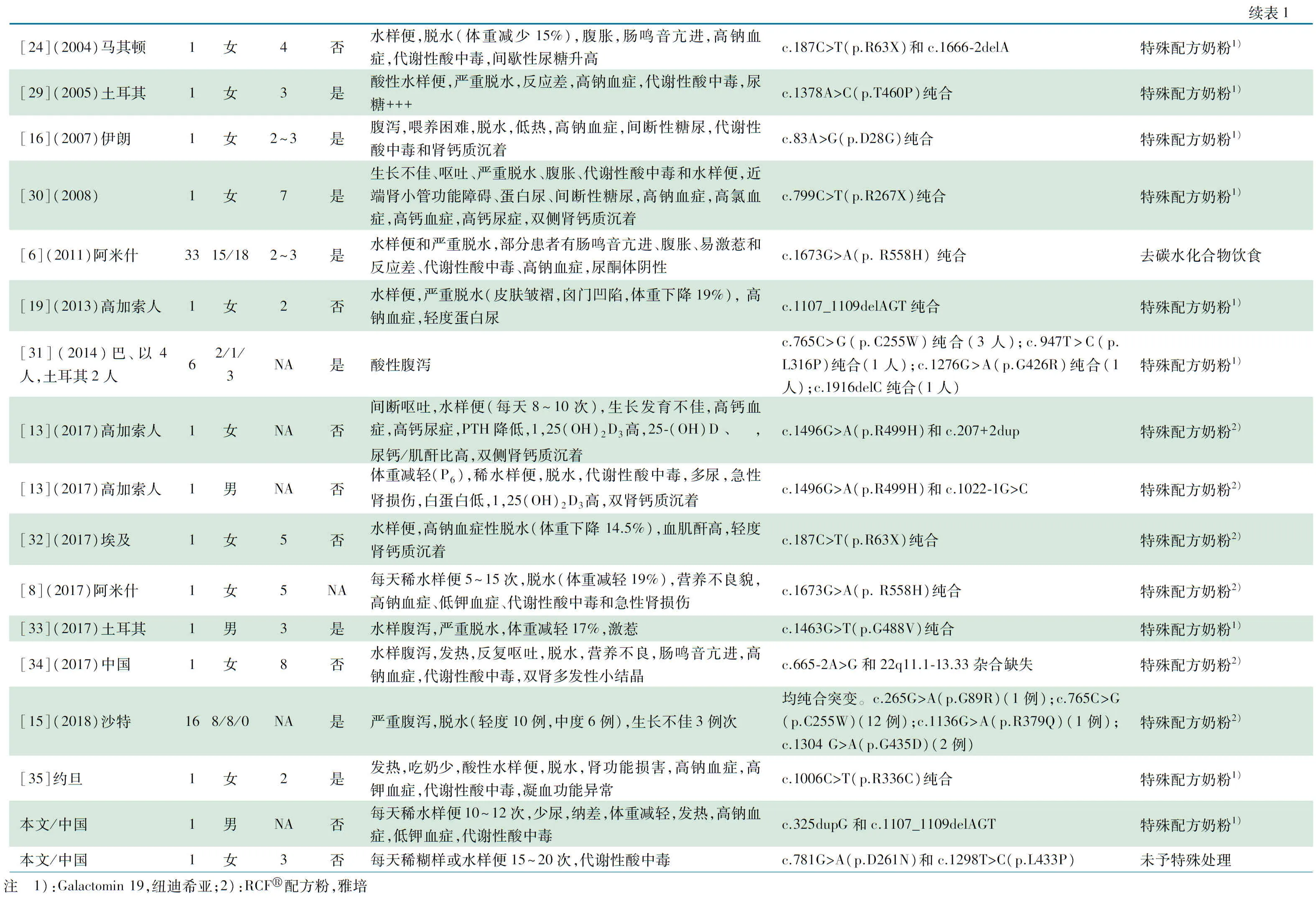
检索PubMed数据库、中国知网、万方和维普数据库,以“(glucose/galactose malabsorption [Title/Abstract])”为英文检索式,以“葡萄糖/半乳糖吸收不良”为中文检索式,检索时间均为建库至2019年5月30日。检索到18篇有临床资料和基因突变分析的文献(中文1篇,英文17篇),与本文合并后共74例患者,详细信息见表2。
74例患者中,男28例,女43例,性别未知3例。46例患者有具体发病年龄记载,其中45例于生后1周内发病,确诊年龄为0~26岁;明确近亲婚配者62例(83.8%)。临床特点:腹泻74例(100%),脱水66例(89.2%),高钠血症11例(14.9%),代谢性酸中毒10例(13.5%),肾损害(尿糖阳性、肾钙质沉着、肾衰竭)24例(32.4%)。治疗:Galactomin 19喂养16例,RCF配方奶喂养5例,52例改为去碳水化合物饮食,仅1例未予特殊处理。预后:存活65例(87.8%),1例(1.4%)因持续肾功能异常、脑积水等继发症状死亡,未提供信息8例(10.8%);随访能耐受少量碳水化合物饮食34例(46.6%),肾钙质沉着5例(6.8%)。66例(89.2%)携带纯合变异(错义变异62例,终止变异2例,缺失变异2例),7例(9.6%)携带复合杂合变异,1例(1.4%)存在包含SLC5A1基因的大片段杂合性缺失。
4 讨论
腹泻是新生儿期较为常见的消化道症状。病因复杂,可由多种因素和病原所导致。近年来随着临床遗传学的进展,逐渐认识到遗传相关疾病可以在新生儿期出现反复腹泻,如免疫缺陷病及代谢性疾病等,其中较为常见的为IL10RA缺陷[2]。葡萄糖/半乳糖吸收不良症(GGM, OMIM 606824)是一种罕见的代谢性疾病,由于葡萄糖和半乳糖吸收不良引发严重的水样腹泻和高渗性脱水,严重者甚至死亡[3]。其致病基因为SLC5A1,定位于染色体22q12.3,迄今已报道了50余种不同的基因突变,不过仍缺乏明确的基因型-表型相关性研究。
GGM是由于小肠黏膜刷状缘表面钠依赖的葡萄糖转运体1结构和功能缺陷造成的,葡萄糖和半乳糖无法在肠道正常吸收,堆积在肠腔内产生大量乳酸,导致酸性水样便,继而引起高钠血症、高渗性脱水、代谢性酸中毒和营养不良,若不及时治疗可能死于低血容量休克[4]。患儿还可能出现腹胀、呕吐、肠鸣音亢进、高氯血症、高钙血症、肾性糖尿和肾钙质沉着症等其他症状。该病最早由Lindquist等[5]于1962年发现并报道,迄今全球范围内已报道的病例300多例。目前仍缺乏相应的流行病学资料,不同人群的发病率存在差异,研究表明该病在阿米什和阿拉伯等近亲结婚率高的地区高发[6, 7],表明GGM呈常染色体隐性遗传模式。本文2例患儿开奶后很快出现稀水样便,伴高钠血症、脱水,例1尤为典型。例1入院时可见营养不良貌,体重仅2 320 g,比出生时减少了26%,血气分析提示合并有低钾血症、高氯血症、代谢性酸中毒等,予禁食并及时予补液、纠酸等支持治疗,电解质紊乱明显改善,先后尝试以蛋白深度水解无乳糖配方奶及氨基酸特殊配方奶喂养,但是随着奶量增加大便次数逐渐增多,血钠亦有上升趋势。粪便有酸臭味,多次查pH值偏低,提示肠内产酸物质增加。患儿病程中反复低热,但CRP一直在正常范围内,可能为糖吸收障碍的不良反应。例2因腹泻就诊,换无乳糖配方奶喂养后大便次数仍较多,且哥哥幼时有相似的临床表现,添加辅食后有所好转。
尽管GGM患儿通常生后2~5 d即出现腹泻症状,但该病极为罕见,临床表现也相当复杂,给早期诊断带来一定的困难。研究表明目前已诊断的病例平均确诊年龄为4.5个月[8]。患儿出现长期水样便、代谢性酸中毒、高钠血症和高渗性脱水,且常规治疗无效时考虑GGM可能[9]。GGM的初步诊断基于以下标准:①出生后不久即腹泻;②粪便特征:包括电解质、pH降低、还原性物质阳性和脂肪等;③无乳糖和氨基酸奶粉不能有效改善症状;④换不含葡萄糖和半乳糖的奶粉喂养时腹泻明显改善,换回原奶粉后再次腹泻;⑤排除感染等疾病。常见的可引起慢性腹泻的其他疾病有:分泌性腹泻,如先天性氯化物腹泻、先天性失钠性腹泻等;细菌和病毒感染;脂质运输障碍,如乳糜微粒潴留性疾病、低脂蛋白血症和脂蛋白缺乏症;胰腺功能不全,如囊性纤维化、Shwachman-Diamond综合征、Johanson-Blizzard综合征和先天性脂肪酶或肠激酶缺乏症。确诊则需依赖以下几种手段:①氢呼气试验,口服葡萄糖或半乳糖(2 g·kg-1)后收集4 h内呼气中的氢气,终浓度峰值超过基础氢值20 ppm以上[10],在婴幼儿中难以实施;②单糖激发试验:口服葡萄糖和半乳糖后血糖反应曲线平坦,而口服果糖和木糖后血糖明显上升[11],结果受多种因素影响,可能出现假阳性或假阴性;③十二指肠组织活检和双糖酶活性检测:结果较可靠,但具有侵入性;④遗传学诊断:通过高通量测序或Sanger测序检测到SLC5A1基因的纯合或复合杂合突变有助于明确诊断,结果为阴性或仅检测到单个杂合突变时,还应考虑SLC5A1基因缺失/重复可能,该方法的应用日趋广泛。
GGM是一种罕见的常染色体隐性遗传疾病,致病基因SLC5A1包含15个外显子,编码由664个氨基酸残基组成的钠/葡萄糖协同转运蛋白[3]。该蛋白是含14个跨膜结构域的跨膜蛋白,分布于小肠和近端肾小管,能促进葡萄糖、氨基酸、维生素及其他离子在小肠和肾的转运[12]。SLC5A1蛋白合成减少或活性降低,均会导致细胞膜转运葡萄糖和半乳糖功能异常,大量未消化的糖和电解质进入结肠,引起渗透性腹泻和严重脱水[4]。这种水样便有时甚至被误认为是多尿[13]。基因检测还能有效地鉴别诊断先天性吸收不良性腹泻(NEUROG3突变)、先天性乳糖酶缺陷(LCT突变)等疾病[14]。截至2019年5月30日,HGMD已收录了SLC5A1基因的50余种突变,包括37个错义突变,5个无义突变,3个剪接位点突变和11个插入/缺失突变,但目前仍缺乏明确的基因型-表型相关性研究。纳入本文检测到的3个可疑致病变异,分析结果表明这些变异分布于除exon7以外的所有外显子,此外,与既往报道一致[20],并未发现变异类型与性别、发病年龄和疾病严重程度之间存在明显的关联。文献报道阿米什家系中33例患者携带相同纯合突变,但其中3例仅表现为慢性腹泻,因而一直未明确诊断[6]。来源不同家庭但携带相同变异的患者其病情严重程度也存在较大差异[15]。此外,错义变异的杂合携带者婴儿期也可能会有腹泻和生长发育不佳[16]。SLC5A1基因突变产生截短蛋白,或使蛋白质构象发生改变,蛋白不能表达到细胞膜上,进而导致转运功能缺陷[3, 17, 18]。本文例1检测到的c.325dupG移码变异导致氨基酸编码提前终止,对蛋白质的结构和功能等均有较大的影响,保守性分析发现另外3个变异位点在各物种间均具有极强的保守性。c.1107_1109delAGT是已收录在HGMD数据库的致病突变,该位点位于连接第8和第9个跨膜区的细胞外襻环处[19]。本文例2及其哥哥检测到的c.781G>A(p.D261N)变异在ExAc数据库仅有2条杂合记录,天冬氨酸为带负电的氨基酸,而天冬酰胺为极性中性,电荷的改变可能导致氨基酸之间的相互作用发生变化,改变蛋白质的折叠和空间结构,Polyphen-2预测为可能有害变异,SIFT和MutationTaster预测为有害变异;c.1298T>C(p.L433P)变异在公共数据库和内部数据库均未见报道,软件预测均为有害变异,亮氨酸和脯氨酸虽同为非极性疏水性氨基酸,但其等电点不一样,突变后的氨基酸体积也较小,该位点位于跨膜区,变异后可能会影响蛋白质与质膜的接触,降低蛋白活性。2例患儿双亲均为杂合携带者,无相关临床表型,患儿为复合杂合突变,符合家系共分离。
GGM的治疗以饮食控制为主,应避免摄入葡萄糖和半乳糖(包括含葡萄糖的口服补液溶液),可以在饮食中添加果糖以补充能量和碳水化合物[20]。另外,应在营养师的指导下适当补充钙和维生素D,减少营养不良风险。目前国外在售的特殊配方奶主要有含果糖的Galactomin 19(纽迪希亚)和去碳水化合物RCF配方奶(雅培)。本文例1入院时体重较出生体重减少830 g(< P3),结合临床表现和实验室检查明确诊断后改用Galactomin 19配方奶喂养,并添加维生素D,腹泻迅速缓解,体重快速增长到3 300 g。该病预后良好,只要及早干预,患儿的生长发育能逐渐趋于同龄儿。Xin等[6]发现部分患者成年后能很好地摄入含碳水化合物的常规饮食,尽管所需时间和改善程度因人而异,但潜在的机制尚不清楚。有学者提出随着肠道菌群的建立,肠道对碳水化合物的耐受力有所改善[21],也有学者发现嗜酸乳酸杆菌可能提高肠道对碳水化合物的耐受力[6]。在营养师的指导下,1岁后患儿可逐步添加低碳水化合物、高果糖含量的饮食,同时监测其大便频率、性状以及其他胃肠道症状[20]。若能耐受,可尝试添加高碳水化合物的水果和蔬菜。然而,也有一些人终身不耐受葡萄糖和半乳糖,甚至不能食用淀粉含量高的蔬菜,主要依赖脂肪、蛋白质和果糖提供能量,因此需要密切关注其肾脏和心血管功能[22, 23]。已有文献报道GGM患儿可能合并有高钙血症、肾钙质沉着症或肾结石[24],而成人中的研究表明高果糖饮食可能会对肾功能产生损害。本文例1住院期间查肾脏B超提示结晶可能,在今后的随访中更应提高警惕。
GGM的罕见性和非特异性临床表现使该病诊断较为困难,而早期诊断和有效管理能预防患儿死亡。分子遗传学检测已被证明是有效的明确诊断手段。本文通过高通量测序技术确诊两例GGM患儿,补充和丰富了SLC5A1基因的突变谱,为临床干预和治疗提供了充分的理论依据,也为未来提供遗传咨询,进行产前筛查、PGD或携带者检测提供了基础。
