硫酸氢酯类药剂浮选铜的量子化学研究
2019-12-06慕红梅马海涛成莉燕张建辉李文雅
慕红梅, 马海涛, 成莉燕, 张建辉, 李文雅
(1.兰州资源环境职业技术学院 环境与化工系, 兰州 730021; 2.兰州市第三十四中学 物理教研室, 兰州 730050)
1 引 言
矿产资源是人类赖以发展的物质基础. 我国虽矿产资源丰富, 但随着开发规模扩大, 形势越来越严峻, 而我国的经济发展对矿物材料仍持有高位需求, 解决经济发展与矿产资源紧缺之间的矛盾已成为一项新的挑战[1]. 矿物浮选法是极为重要的矿产资源开发手段, 浮选捕收剂的品种和质量直接关系到浮选工艺发展效果的优劣, 而浮选药剂大都对环境有不同程度的污染, 大规模试验并不科学. 因此, 针对特定的浮选任务, 设计出新型高效、廉价、环保的浮选药剂, 对我国矿物的综合回收利用以及生态环境安全具有重要意义. 开发新型浮选药剂的方法有物理化学方法、拓扑学方法和分子模拟方法[2,3]. 近年来发展比较迅速的分子模拟方法包括量子化学方法和分子力学动力学方法, 采用分子模拟技术可以降低浮选新药剂开发的经济成本, 保护环境, 提高设计效率, 这对于现存的复杂难选矿物的开发利用具有重大的理论指导和实际应用价值.
近年来, 国内外运用基于密度泛函理论的量子化学方法研究设计浮选药剂主要集中在黄铁矿和黄铜矿的分子结构[4,5]、黄药[6,7]、胺类[8]等浮选药剂. 未有关于硫酸氢酯类浮选药剂的相关研究报道. 本研究选用15~21个碳原子的硫酸氢酯, 研究其几何参数、电荷密度分布及与铜离子吸附后能量变化. 通过以上研究能为浮选药剂设计提供理论依据和设计方法.
2 计算方法
采用密度泛函(DFT)B3LYP方法[9,10], 6-311+G(d, p)基组[11], 对碳原子数为15~21的硫酸氢酯进行参数优化, 得到各物质稳定结构、 能量和电荷分布. 进一步计算了各物质与铜离子作用后的稳定结构和吸附能. 全部计算工作采用Gaussian09程序完成[12], 分子的几何构型全部由GaussView 程序从计算结构直接转换生成, 见图1.

图1 C15H32O4分子结构图Fig. 1 The diagram of molecular structure of C15H32O4
3 结果与讨论
碳原子数为15~21的硫酸氢酯类浮选药剂离子结构和吸附铜离子后的稳定结构, 见表1.
从表1可以看出, 随着碳原子个数增加, 硫酸氢酯类阴离子结构中碳链出现了弯曲. 当离子吸附铜离子后, 碳链也出现了弯曲, 且弯曲程度全部增大.
3.1 优化后能量变化
前线轨道理论指出, 分子的最高占据轨道(Highest Occupied Molecular Orbital, HOMO)与最低空轨道(Lowest Unoccupied Molecular Orbital, LUMO)决定着分子的电子得失与转移, 从而决定着分子的空间取向和化学反应. HOMO上的电子能量最高, 最活泼, 也最容易失去电子, 还原性强,即HOMO 值越大, 越容易失电子, 还原性越强; 而LUMO上的电子能量最低, 最稳定, 最容易

表1 分子结构图
接受电子,具有氧化性, 即LUMO值越大, 越容易得电子, 氧化性越强[13]. DFT方法计算的碳原子数为15~21的硫酸酯类分子的前线轨道能量参数见表2, 其中n为碳原子个数,ELUMO为最低空轨道能量,EHOMO为最高占据轨道能量,ΔE1=ELUMO-EHOMO为前线轨道能量差值, 即能隙.

表2 硫酸氢酯的能量对比
按照化学反应性的前线分子轨道理论, 反应物的能隙是一个重要的稳定性指标,ΔE值越大, 反应物稳定性越高, 反应中活性越低, 而ΔE值小则意味着反应物易给出电子, 具有高的反应活性[3,14]. 碳原子数为15~21的硫酸氢酯类药剂中ΔE均为正值, 该酯在吸附铜离子时都存在电子转移, 其中C17的反应活性较强, C19~C21反应活性较低.
3.2 电荷密度分布变化
碳原子数为15~21的硫酸氢酯相同位置上的核心原子的电荷密度分布列于表3中, 吸附铜离子后, 核心原子的电荷密度分布列于表4中. 从表3可知, 在与铜离子吸附前, C15~C17硫酸氢酯相同核心原子上的电荷密度相同, 说明这三种酯对铜离子的吸附作用相同, C18~C21硫酸酯中相同核心原子上的电荷密度发生变化, 说明随着碳原子数的增多, 酯对铜离子的吸附作用逐渐变弱[15], 与分子结构和前线轨道能量变化一致.
从表4可知, 吸附铜离子后, 电荷密度分布出现明显变化, 相同碳原子数的硫酸酯, 硫原子和四个氧原子及和氧原子相连的碳原子上的电荷密度都出现了较大的改变. 从电荷密度分布可知, 6O上的电荷密度最小, 最容易被铜离子进攻, 即为铜离子的最佳吸附位置, 铜离子的电荷也随着碳原子数增大而逐渐减小.
表3 硫酸氢酯核心原子的电荷分布
Table 3 The distributions of charge in the core atoms of hydrogen sulfate esters

Number1S5O6O2O4O7C10C152.435-0.965-0.986-0.986-0.772-0.834-0.379162.435-0.965-0.986-0.986-0.772-0.834-0.379172.435-0.965-0.986-0.986-0.772-0.834-0.379182.434-0.963-0.990-0.984-0.771-0.831-0.379192.434-0.963-0.990-0.984-0.771-0.831-0.379202.434-0.963-0.990-0.983-0.771-0.830-0.379212.433-0.963-0.990-0.983-0.771-0.830-0.379
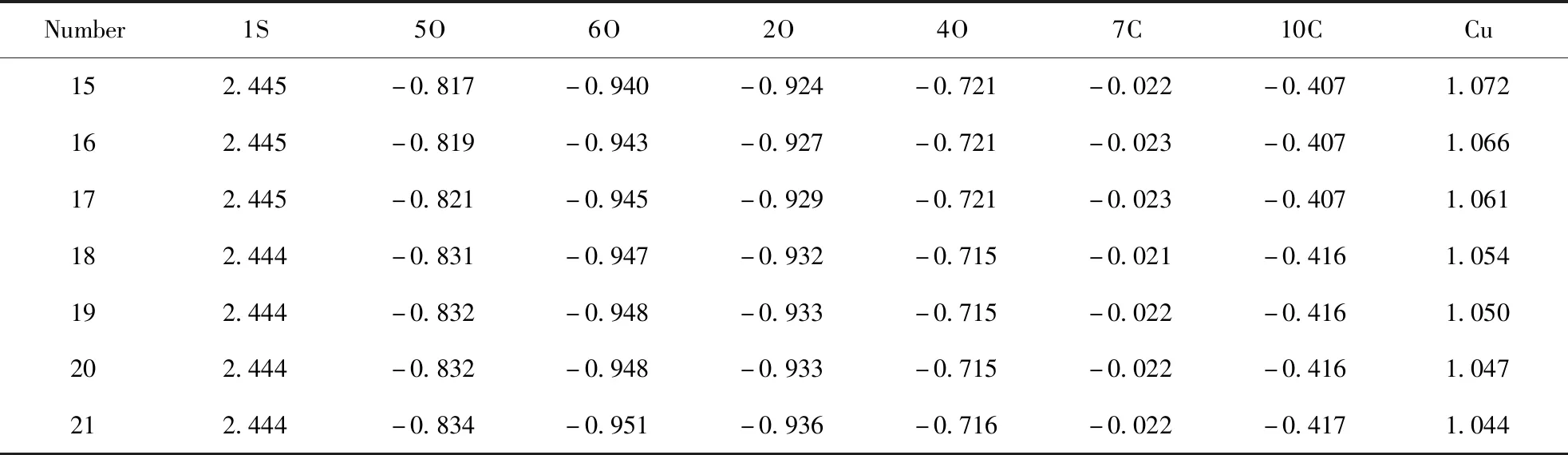
表4 硫酸氢酯吸附铜离子后核心原子的电荷分布
3.3 吸附能变化
碳原子数为15~21的硫酸氢酯与铜离子的吸附能变化列于表5中, 在ΔEabs=ECn-Cu-ECn-ECu中,ECn为不同碳原子数的硫酸氢酯的能量,ECu为铜离子的能量,ECn-Cu为不同碳原子数的硫酸氢酯与铜离子作用的吸附能.

表5 硫酸氢酯与铜离子作用的吸附能
从表5可知,ΔEabs均为负, 碳原子数为15~21的硫酸氢酯均可以与铜离子发生化学吸附, 且硫酸氢酯吸附铜离子后, 总吸附能逐渐降低.
4 结 论
采用密度泛函DFT-UB3LYP方法, 6-311+G(d, p)基组, 对碳原子数在15~21的硫酸酯类浮选药剂吸附铜离子进行了计算并得出以下结论:
(1)从前线轨道能隙来看, 碳原子数为15~21的硫酸氢酯类浮选药剂中C17的反应活性最强, C19~C21反应活性较差;
(2)从电荷密度分布可知, 6O电荷密度最小, 是最容易被铜离子进攻的位置, 即为铜离子的最佳吸附位置;
(3)从总吸附能的计算可知, C18~C21结构较为稳定, 这与前面的电荷计算结果一致.
通过计算, 综合考虑各种因素C17(十七烷基硫酸氢酯)浮选效果较好.
