Tumor heterogeneity of acute myeloid leukemia:insights from single-cell sequencing
2019-11-02AiLiChenShoYnHuQinFeiWng
AiLi Chen, ShoYn Hu, Qin-Fei Wng,c,*
aCAS Key Laboratory of Genomic and Precision Medicine, Collaborative Innovation Center of Genetics and Development, Beijing Institute of Genomics, Chinese Academy of Sciences, Beijing, China; bDepartment of Hematology and Oncology, Children's Hospital of Soochow University, Suzhou, China; cUniversity of Chinese Academy of Sciences, Beijing, China
Abstract
Keywords:Acute myeloid leukemia, Heterogeneity, Single-cell sequencing
1.INTRODUCTION
Peter Nowell first proposed that the formation of tumors is an evolutionary process1and that genetic mutations are traditionally recognized as molecular determinants of this process.Although unique combinations of mutations are commonly used to define different subclones, it is now widely accepted that genetic and epigenetic alterations collectively contribute to functional heterogeneity, such as self-renewal and drug resistance within individual tumors.2Intratumoral heterogeneity is a key determinant in disease progression and treatment response;therefore, a comprehensive analysis of tumor heterogeneity has important clinical implications and may provide valuable insights for the development of anticancer therapies.3
Acute myeloid leukemia (AML) is one of the best studied cancer type owing to the easy accessibility of samples and welldefined cellular hierarchy in the normal hematopoietic system.4This unique feature with the relatively low recurrent somatic mutation rate of the disease5make AML an excellent model to explore intratumoral heterogeneity and clinical significance.6Laboratory-based technologies such as fluorescence in situ hybridization and flow cytometry as well as high-throughput next-generation sequencing (NGS) have revealed that AML comprises heterogeneous subpopulations characterized by distinct genetic and epigenetic alterations.7,8However,the multiple layers of heterogeneity have remained ambiguous until the introduction of single-cell sequencing(SCS).SCS has empowered the cancer field to achieve an unprecedented understanding of the molecular basis of intratumoral heterogeneity.In this review,we summarize how SCS technology contributes to our understanding of AML heterogeneity,with a particular focus on insights that are only revealed from single-cell analyses (Table 1).
2.USING SINGLE-CELL GENOME SEQUENCING TO UNAMBIGUOUSLY DEFINE THE GENETIC HETEROGENEITY OF AML
AML is thought to originate from a subset of hematopoietic stem/progenitor cells(HSCs/HPCs).9,10By gradually accumulating mutations and numerous additional genetic and epigenetic abnormalities, the found cells take a complex trajectory path to finally evolve into functionally diversified subpopulations.11-13Obtaining genomic information at the single-cell level is the ultimate solution to dissect clonal diversity.To achieve this goal,current technology requires whole-genome amplification(WGA)of DNA extracted from an individual cell to generate enough DNA for sequencing.WGA methods continue to face several technical limitations,such as a high allele dropout rate,high falsepositive rate, and chimera formation, which have been well described in other reviews.14,15Currently, most single-cellgenomic studies of AML have relied on genotyping of somatic mutations pre-detected by genome sequencing of a mixed cell population, with the genotyping libraries constructed from single-cell WGA or colony-forming units derived from a single cell.16-19
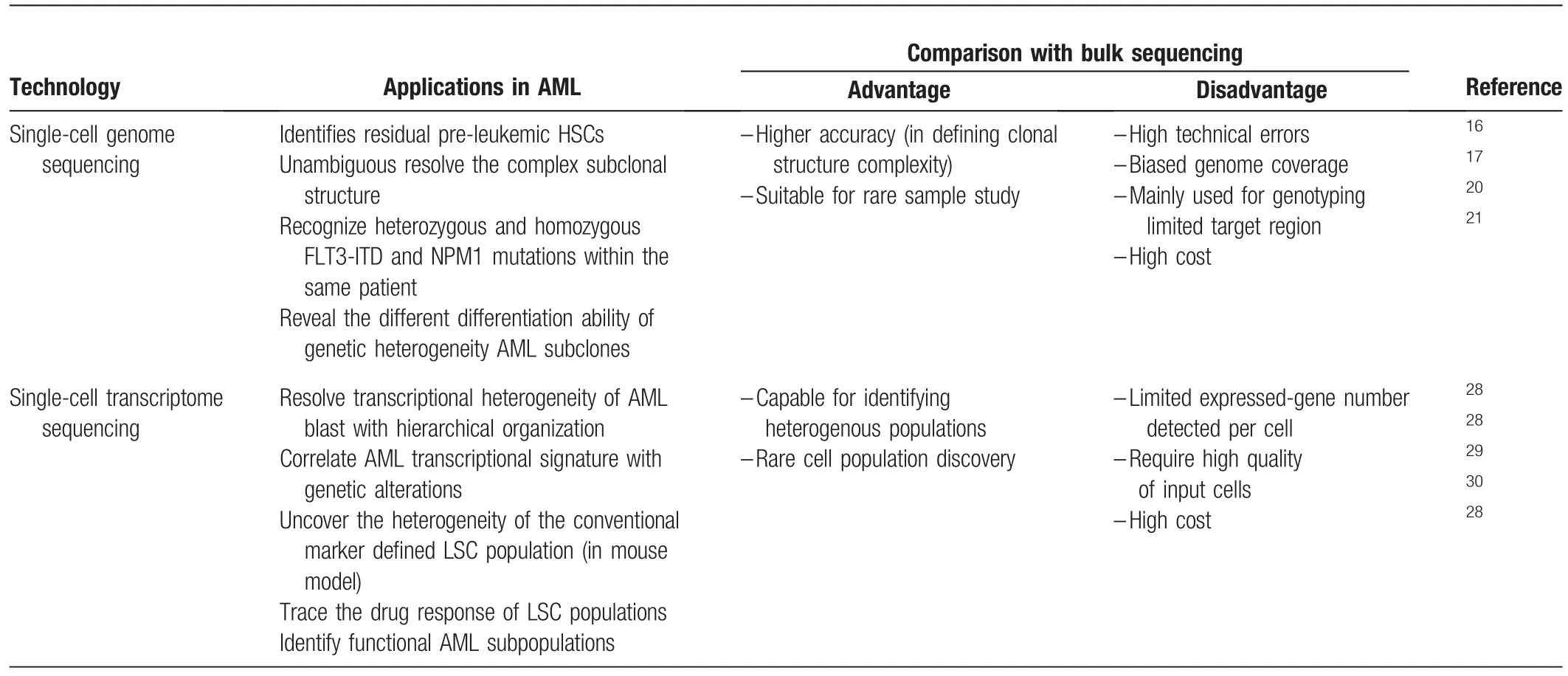
Table 1 Applications of Single-cell Sequencing technologies in AML studies.
Single-cell genotyping of limited somatic mutations (usually<10) in a small number of cells (several to several hundreds)allows the determination of a relatively clearer clonal structure of de novo AML compared with NGS of bulk samples.AML was originally viewed as evolving from a linear accumulation of multiple driver mutations.However, the estimates of subclone size in deep bulk sequencing indicate that AML is likely evolved with a complex branching architecture.6Despite this improved understanding, it remains difficult to determine the exact evolutionary trajectory and clonal structure based on the variant allele frequency (VAF), particularly when the VAFs of two subclonal mutations are similar.Single-cell DNA sequencing can resolve the combined pattern of multiple mutations in an individual cell and subsequently provide evidence for either linear or branched structures in individual AMLs.Remarkably,singlecell genotyping of pre-detected mutations identifies residual preleukemic HSCs that exist in bulk AML cells and constitute a cellular reservoir with linearly accumulated founder mutations.These pre-leukemic cells are predisposed to acquire additional mutations and evolve into a distinct clonal hierarchy observed in the full-blown disease.16Single-cell gene capture of 1953 somatic single nucleotide variants (SNVs) from patients with AML unambiguously demonstrates the origin of two separate subclones by detecting mutually exclusive SNVs,which represent an evolutionary branch point from the founding clone.17Interestingly, most VAF in bulk sequencing is calculated under the assumption that all mutations occur heterozygously in single cells, and a study using single-cell genome analysis has revealed the existence of both heterozygous and homozygous FLT3-ITD and NPM1 mutations in an individual patient.20These data suggest that bulk sequencing overestimates the number of leukemic cells but underestimates the clonal diversity of the blast.In an intriguing study, single-cell genotyping was performed on flow-sorted AML blasts and mature myelomonocytes.These two cell populations contain the same founding clone but distinct subclones,indicating that genetic heterogeneity impacts the differentiation ability of AML subclones.21A recent study adopted droplet microfluidics for single-cell DNA genotyping, which enabled a comprehensive analysis of thousands of AML cells in parallel.22This single-cell DNA sequencing has potential in the development of high-throughput genomewide detection of genetic alterations in the future.
These single-cell genomic studies16-20strongly support the clonal architecture inferred from the analysis of bulk samples.Importantly,analyses of AML at single-cell resolution unambiguously resolve the subclonal structure that cannot be appropriately assigned through mutation frequencies or unknown mutation status (heterozygous or homozygous).Thus, these studies reveal the previously unappreciated complexity of the clonal structure of AML.
3.USING SINGLE-CELL TRANSCRIPTOME SEQUENCING TO REVEAL THE FUNCTIONAL HETEROGENEITY OF AML
The transcriptome is the dynamic output of all genetic information, which closely reflects the functional status of cells.Compared with genomic analyses, single-cell transcriptome sequencing (scRNA-seq) is a more developed technology that allows for a high-throughput(several hundreds to ten thousands cells)detection of tens of thousands of transcripts.Considerable technical progress has been made over the past years, including the introduction of the Moloney murine leukemia virus, which has template-switching activity to ensure amplification of fulllength mRNA transcripts,23and the development of unique molecules index for reduced amplification bias.24A commercial provider(10×Genomics®,7068 Koll Center Parkway,Suite 401,Pleasanton, California 94566) ensures the widespread use of high-throughput SCS technology.The scRNA-seq of a normal hematopoietic cell population confirms the structured hierarchy of a normal blood development of stem cell derived-myeloid and-lymphoid lineages,25,26and more importantly, leads to the discovery of a novel dendritic cell(DC)subset.27Transcriptomic profiling of AML has significantly improved our understanding of tumor hierarchical structure and led to the identification of novel cell types, leukemic stem cells (LSCs), and drug-resistant subpopulations.28-30
Unique leukemia-enriched plasma membrane proteins (e.g.,CD25 and IL1RAP) enabled the prospective isolation of two AML blast populations from individual patients,and the separate subclones were shown to be genetically and epigenetically distinct and to have different functions in their drug sensitivity, growth,and engraftment behavior.31These data demonstrate the existence of functional diverse subpopulations within individual AMLs.scRNA-seq data were used to profile 16 patients with AML and 5 healthy donors and to classify AML cells into 6 subpopulations along the HSC to myeloid axis of the normal cell hierarchy.Each population was characterized by a unique expression signature, including HSCs, progenitors, granulocytemacrophage progenitors (GMPs), promonocytes, monocytes,and conventional DCs.Such signatures exhibit a close correspondence to the genetics of the AMLs.For example, TCGA tumors with high GMP-like and promonocyte-like scores derived from scRNA-seq perfectly overlap with PML-RARA fusions.Different AML cell signatures are further correlated with clinical outcomes such the outcomes are better in patients with a higher GMP-like score than in those with a higher HSC/Progenitor-like score.28The molecular classification of AMLs has also been explored by mass cytometry and chromatin accessibility at the single-cell level.By comparing surface and intracellular signaling proteins simultaneously in healthy and leukemic cells by singlecell mass cytometry,Levine et al32have reported that the tightly coupled surface and signaling phenotypes in healthy cells were perturbed in AML cells.Compared with cell surface markers,the signatures of intracellular signaling proteins are more reliable to predict clinical outcomes in patients.32The analysis of single-cell chromatin accessibility also reveals the regulatory heterogeneity of AML cells resembling a normal blood cell hierarchy, with a similar pattern distributed along the HSC to myeloid axis.33These studies indicate an intrinsic relationship among the transcriptomes, proteomes, and epigenomes in AML cells.
AML LSCs have been recognized as the apex of the AML cellular hierarchy within the heterogeneous population, with properties of self-renewal, cell cycle quiescence, and chemoresistance.34It is believed that a cure for AML depends on eradicating the LSCs.However,no definitive phenotypic markers are known to purify LSCs.Intriguingly, the LSC-enriched population can be recognized by characteristic transcriptional signatures,such as the“LSC score.”35The use of the LSC score in a single-cell classification can help identify and trace the LSC population during treatment and provide opportunities to improve identification of the LSC population.Indeed, the LSC expression signature has been applied in single-cell analysis of the MLL-AF9 mouse model and has uncovered the heterogeneity of the conventional marker-defined LSC population (Lin-Il7r-Kit+Sca1-CD34+CD16/CD32+)that contains two independent selfrenewing lineages with different clonal activities.29In contrast,LSCs have metabolic features that require active oxidative phosphorylation status.36scRNA-seq has been used to trace the response of the LSC population during venetoclax with azacitidine treatment, which can disrupt the energy metabolism in AML patients.30Interestingly, SCTS also leads to the identification of a differentiated monocyte-like AML cell population, which suppresses T-cell activation in vitro and might have immunomodulatory functions that contribute to the pathogenesis of the disease.28
4.CONCLUSION: LIMITATIONS AND PROSPECTIVE
Multiple SCS technologies have been applied in cancer research and have achieved remarkable progress.However, significant technical improvements are needed to achieve a comprehensive view of tumor heterogeneity.First,SCS technology is still limited by amplification bias and extensive technical errors,particularly in single-cell genome sequencing.37Second, the need for simultaneous acquisition of multiomics data of a single cell has not been fulfilled.For example,the co-existence of leukemic,preleukemic,and normal blood cells in AML samples requires the integration of mutational spectrum and transcriptional states to optimally classify the functional heterogeneous subpopulations.To date, most attempts at systematically detecting both genetic and RNA expression information from the same single cell are either expensive or have low sensitivity,38-40thus restricting the widespread use of integrative analysis.However, some limited success has been achieved with a relatively low throughput in the studies of hematopoietic malignancies.These include single-cell transcriptome analyses in combination with successful detection of the BCR-ABL breakpoint in chronic myeloid leukemia41and a novel method that allows mutational analysis(up to 12 different mutations) and parallel RNA sequencing from the same single cell in myeloproliferative neoplasm.42Furthermore,efforts have been made to combine transcriptome detection with surfacemarker protein analysis43or assay for transposase-accessible chromatin using sequencing44within a single cell.These studies hold promise for the future development of multiomics integrated at the single-cell level.
Dissecting AML heterogeneity using SCS has provided novel insights into the molecular mechanisms underlying drug resistance and disease progression.Single-cell targeted mutation analysis in AML treated with the FLT3 inhibitor quizartinib has revealed highly complex drug-resistance clones at relapse,demonstrating marked clonal complexity associated with therapeutic resistance.45By comparing clonal structures identified by single-cell DNA sequencing at diagnosis and relapse,multiple bypass pathways have been identified in patients with AML having IDH2 mutations that can potentially be targeted for treatment.46
While these studies serve as excellent examples of how new knowledge from SCS can aid in the development of new drugs and treatment options, dissecting intratumoral heterogeneity holds a relatively greater potential for clinical application in the era of precision medicine.For example, new biomarkers identified from previous unknown drug-resistant subpopulations could be used to better stratify patients with AML.Moreover,SCS in combination with a more sensitive targeted mutation detection method can be used to monitor functionally diverse subclones with different drug-sensitivities and stem cell potential during treatment and disease progression.This information will be invaluable in improving prognostic prediction and detecting minimal residual disease.With rapid development of these advanced technologies, it is conceivable that SCS will have a tremendous impact on the clinical practice of AML.
杂志排行

血液科学的其它文章
- Successful ex vivo expansion of mouse hematopoietic stem cells
- Cell cycle regulation and hematologic malignancies
- Will immune therapy cure acute myeloid leukemia?
- Engineered human pluripotent stem cell-derived natural killer cells: the next frontier for cancer immunotherapy
- Hematopoietic stem cell metabolism and stemness
- Epigenetic regulation of hematopoietic stem cell homeostasis