The role of cholesterol metabolism in leukemia
2019-11-02LingZhoHuienZhnXinyJingYngqiuLiHuiZeng
Ling Zho, Huien Zhn, Xiny Jing, Yngqiu Li, Hui Zeng,*
aDepartment of Hematology, Xiangya Hospital, Central South University, Changsha, Hunan, China; bDepartment of Hematology,The First Affiliated Hospital of Jinan University, Jinan University, Guangzhou, Guangdong, China; cInstitute of Hematology, Jinan University, Guangzhou, China.
Abstract
Keywords:Cholesterol metabolism, Drug resistance, Leukemia, Mevalonate pathway, Statins
1.INTRODUCTION
Leukemia is a malignant clonal hematopoietic disease characterized by abnormal aggregation of immature blasts in the bone marrow and hindrance to the formation of normal blood cells.On the basis of the involved cell type and expansion speed,leukemia is classified into four major types,namely acute myelogenous leukemia (AML), acute lymphocytic leukemia(ALL), chronic myelogenous leukemia (CML), and chronic lymphocytic leukemia (CLL).Even rapid progress in the discovery of novel drugs and the development of new approaches, however, highly effective treatments with minimal side effects are still in shortage.Consequently,there is an urgent need to elucidate the mechanism of leukemia-initiating,developing, relapsing and drug resistance, and further screen novel targets.
The metabolic profiles in leukemia cells were found to be distinct from the normal process.Chen et al reported that a total of 47 metabolites and 45 metabolic pathways were altered in AML patients,1among which glucose and lipid metabolic alterations were notable.In another study, it was reported that cholesterol metabolism was aberrant in both ALL and AML patients.2
Cholesterol is indispensable for cancer cells to achieve fast proliferation as it functions in several biological aspects.Apart from acting as an essential structural component for cell membrane,3cholesterol also provides a platform for cell signal transductions by way of lipid raft element, which are master regulators of cell fate.4Moreover,the intermediate metabolites in cholesterol synthesis regulate the post-translational prenylation of many proteins, including small GTPases, which is a vital process to make those proteins function normally.5
It has been demonstrated that leukemia cells display increased synthesis and uptake of cholesterol to satisfy their rapid growth,as indicated by elevating 3-hydroxy-3-methylglutaryl coenzyme A (HMG-CoA) reductase (HMGCR) activity and expression,and enhancing low-density lipoprotein receptor (LDL-R) mediated degradation of LDL.6-8With it comes extensive attention drawn by cholesterol modulators nowadays, many efforts have been made to test the therapeutic efficacy and promote clinical translation.For example,SWOG S0919,a Phase 2 trial,revealed an encouraging complete remission rate in relapsed AML patients treated with high dose pravastatin combined with idarubicin and cytarabine.9
In this review, we provide an outline of the cholesterol homeostasis, as well as discuss the function of cholesterol and its derived metabolites in leukemia expansion, progression, and drugresistance.Wefurtherbrieflyhighlightthe researchprogresses of diverse inhibitors targeting cholesterol metabolism in leukemia,which may open a new therapeutic window for treating leukemia.
2.CHOLESTEROL METABOLISM NETWORK
Cholesterol is both synthesized and taken up by receptormediated endocytosis (Fig.1).Cells delicately balance the importation and biosynthesis of cholesterol by a tightly regulated feedback of LDL receptors and HMGCR.The brief procedures are as follows.
2.1.De novo cholesterol synthesis
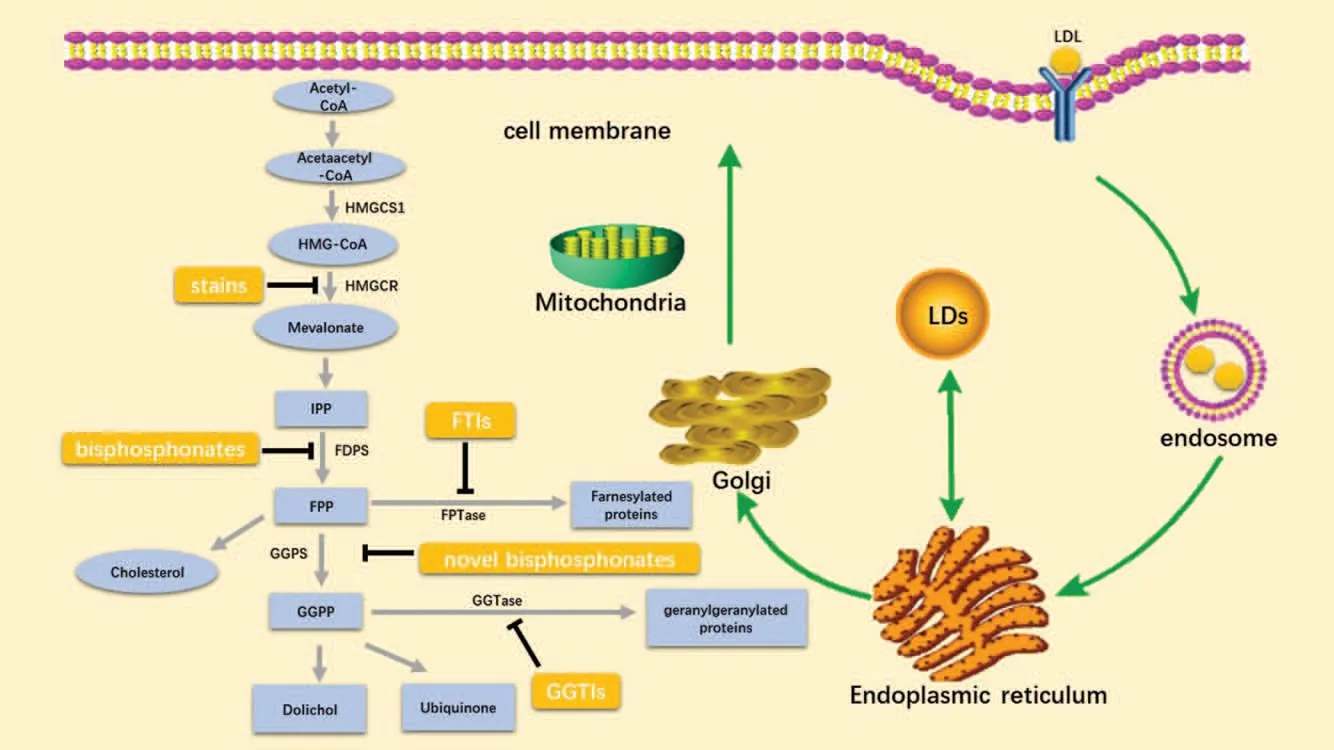
Figure 1.Overview of the cholesterol metabolism and the promising inhibitors.Cholesterol metabolism,which uses acetyl-CoA to produce sterols and isoprenoid metabolites,is indispensable for cancer cells.Cholesterol is both de novo synthesized(the left part)and taken up by receptor-mediated endocytosis(the right part).Key metabolic enzymes discussed in this review are marked in black besides the corresponding arrows,derived intermediate metabolites are shown in light blue boxes, and agents targeting cholesterol metabolism are shown in orange boxes.Many reactions have been omitted for simplicity.HMG-CoA, 3-hydroxy-3-methylglutaryl CoA;HMGCS1,HMG-CoA synthase 1;HMGCR,HMG-CoA reductase;IPP,isopentenyl-diphosphate;FDPs,farnesyl diphosphate synthase;FPP,farnesyl diphosphate;GGPS,geranylgeranyl pyrophosphate synthase;GGPP,geranylgeranyl-diphosphate;FTIs,farnesyl-transferase inhibitors;GGTIs,geranyl transferase inhibitors; LDL, low-density lipoprotein.; LDs lipid droplets.
Cholesterol can be synthesized in the plasm by the mevalonate(MVA) pathway.10,11The MVA pathway leads to the synthesis of sterols and isoprenoids that are shown to be crucial for tumorgrowth.12The MVA pathway begins with the synthesis of acetoacetyl-CoA from acetyl-CoA.Next,HMG-CoA is produced by the condensation of acetoacetyl-CoA and acetyl-CoA,catalyzed by HMG-CoA synthase 1 (HMGCS1).The next step is the conversion of HMG-CoA to MVA by HMGCR, which is the first committed step of the MVA pathway.MVA is then converted into isopentenyl-diphosphate (IPP) and dimethylallyl pyrophosphate (DMAPP) through a series of enzymatic steps.Farnesyl diphosphate synthase (FDPS) catalyzes sequential condensation reactions of DMAPP with two units of IPP to form farnesyl pyrophosphate (FPP).Geranylgeranyl pyrophosphate synthase (GGPS) catalyzes yet another condensation reaction to form geranylgeranyl pyrophosphate (GGPP).GGPP can be converted into dolichol and ubiquinone (coenzyme Q,CoQ) through multiple enzymatic steps.On the other hand,farnesyl-diphosphate farnesyltransferase 1(FDFT1)converts FPP into squalene,which is then converted into cholesterol through a series of enzymatic steps.
2.2.Receptor-mediated endocytosis
Other than de novo synthesis, the increase of intracellular cholesterol is also triggered by the uptake of LDL.13,14First,circulating LDL particle binds to LDL receptor localized in the cell surface and is engulfed by endocytosis, forming a vesicle(endosome).LDL dissociates from the receptor inside the endosome, and the latter is recycled to the cell surface.Next LDL traffics to the lysosomal system where it is hydrolyzed to free cholesterol and fatty, functioning as the component of the cell membrane and other cell membrane-bound organelles acids.
3.THE EFFECTS OF ABERRANT CHOLESTEROL METABOLISM ON LEUKEMIA
Existing data on intracellular cholesterol levels in leukemia cells are discrepant: the intracellular cholesterol level may be higher15,16or lower17,18in comparison to peripheral blood mononuclear cells(PBMCs)from healthy donors,which may be due to cell differentiation status and different experimental approaches.But it is widely accepted and accordant that the utilization rate of cholesterol is more rapid6,19and cholesterol synthesis is enhanced in leukemia cells.20Moreover, negative feedback repression of leukemia cells is defective and limited in high-sterol media.7Based on available research findings,aberrant cholesterol metabolism primarily has the following effects.
3.1.Promotion of cell growth and inhibition of apoptosis
As a critical component of lipid raft, cholesterol is tightly related to the abundance and function of numerous growth signaling proteins,remarkably affecting the proliferation rate of ALL and CLL cells.16Besides,cholesterol promotes cell viability in subsets of acute leukemias by modulating VEGF:VEGFR-1 signaling, leading to leukemia expansion and extramedullary colonization ultimately.16Drugs that aim at reducing cholesterol would counteract this function.For example, when depressed expression of FDPS and farnesyl diphosphate farnesyltransferase(FDFT1) as a result of 5-aza-2′-deoxycytidine treatment,leukemia cells present with substantially reduced cellular cholesterol and growth inhibition.21The treatment of 2-hydroxypropyl-β-cyclodextrin,a cholesterol modifier,22reduces intracellular cholesterol leading to significant leukemic cell growth inhibition by means of G2/M cell-cycle arrest and apoptosis.23In human myeloid leukemia HL-60 cells,the effect of growth inhibition and apoptosis is triggered by α-tomatine, a glycoalkaloid, and is partially abrogated by the addition of cholesterol.24Moreover, the specific inhibitors of cholesterol,presenting opposite effects, will be discussed in detail subsequently.
3.2.Disturbance with cell differentiation
All trans-retinoic acid (ATRA), as a potent inducer of differentiation,is classically used to treat AML-M3 in the clinic.Cells pretreated by compactin, a competitive inhibitor of HMGCR, obviously enhance the differentiation rate in HL-60 cells.25Cholesterol starvation or treatment with zaragozic acid,which inhibits squalene synthase,induces differentiation of HL-60 cells following a granulocyte lineage.26
3.3.Contribution to drug resistance
Abnormal cholesterol homeostasis is observed in both refractory leukemia and leukemia cells exposed to chemotherapy.27Recent research reported that the HMGCR expression is up-regulated in drug-resistant myeloid leukemia cell lines compared with the parental cell lines.28Likewise,drug-resistant ALL cell lines have increased cholesterol levels.29AML cells prevent themselves against drug cytotoxicity by swiftly boosting protective cholesterol levels via elevating HMGCR and LDL-R mRNA level.20Most leukemia cells depend on de novo cholesterol synthesis to increase cholesterol levels.Nevertheless,a subset of AML cells relies on increased LDL-C enrichment to accumulate cholesterol.20
4.THE ROLES OF DERIVED INTERMEDIATE METABOLITES IN LEUKEMIA
4.1.FPP and GGPP
FPP and GGPP function as the substrates for farnesylation and geranylgeranylation respectively.This process is catalyzed by either farnesyl transferase (FTase) that covalently attaches a farnesyl moiety or geranylgeranyl transferases I and II(GGTase I and II) that covalently attach a geranylgeranyl moiety to carboxyl-terminal cysteines.30
Disordered RAS signal transduction is a major pathogenesis of myeloid leukemias.31As a small GTPases, the RAS family requires mevalonate-derived farnesyl or geranylgeranyl isoprenoid modifications for membrane localization32and further transduces mitogenic signals by activating downstream effectors such as Rafs and PI3K,which regulate proliferation,differentiation, and malignant transformation.33In other words, reducing the production of FPP and GGPP could contribute to the antileukemic activity.For example,depletion of cellular GGPP blocks protein geranylgeranylation of ERK Rac, RhoA, and Rap1,resulting in activation of caspases and pro-apoptotic signaling,and suppressing leukemia growth ultimately.34
4.2.CoQ
CoQ localizes to the inner membrane of the mitochondria,acting as an electron carrier within the mitochondrial respiratory chain,thus enabling ATP production.35CoQ(10)is necessary to control superoxide levels in HL-60 cells.36CoQ analogs,CoQ0,significantly reduced HL-60 cell viability and triggered apoptosis via ROS-mediated up-regulation of the Voltage-dependent anion channel 1 (VDAC1) signaling pathway.37Ubiquinone-based compounds could inhibit human leukemic cell growth by disrupting trans-plasma membrane electron transport (tPMET)and then disturbing intracellular redox homeostasis.38Besides,CoQ(10) has protective effects on anthracyclines induced cardiotoxicity.39
4.3.Dolichol
Dolichol and dolichyl monophosphate(Dol-P)are involved in the attachment of carbohydrate chains to proteins in the formation of N-linked glycoprotein.40Dol-P induces apoptosis in human leukemia U937 cells, while dolichol fails to do so.41During this apoptotic execution, reduction in mitochondrial transmembrane potential(immediately),42increase in membrane fluidity (5-20min),43mitochondrial morphological changes (5-60min),42translocation of apoptosis-inducing factor(AIF)(1-3 h),44increase in caspase-3-like protease activity (2-4h)45and DNA ladder formation (3-4h)46were observed successively.Direct disruption of mitochondrial respiratory complexes and the consequent reactive oxygen species(ROS)generation also play a critical role in the initiation of Dol-P-induced apoptosis.47
5.AGENTS TARGETING CHOLESTEROL METABOLISM
Both cholesterol synthesis and transport are promising targets for leukemia therapy, as researches concentrating on intestinal cholesterol absorption have got certain attention.For example,CML is featured with accumulated cholesteryl ester (CE).Blocking CE with avasimibe could profoundly suppress CML cell proliferation and sensitize resistant cells to imatinib treatment.48However, present researches mainly shed light on cellular cholesterol synthesis.Herein,we will discuss several types of widely studied inhibitors in leukemia (Fig.1).
5.1.Statins
Used extensively as a cholesterol-lowering agent in the clinic,statins inhibit HMGCR by blocking the binding site of HMGCoA.49They are generally classified into two groups,hydrophobic and hydrophilic.Hydrophobic statins (e.g., atorvastatin,simvastatin, lovastatin, fluvastatin) are more effective than hydrophilic statins (e.g., pravastatin and rosuvastatin) in cancer treatment since the former are easier to cross the biological membranes.50
Statins exert cytotoxicity on cancer cells,51ascribing to the inhibition of both cholesterol synthesis and various MVA-derived metabolites.5,52Preliminary clinical investigations have suggested that statins have anti-leukemia activities.27,53,54Even multidrug-resistant (MDR) sublines of myeloid leukemia cell lines also show sensitivity to lovastatin.28Two clinical trials further reveal that statins can improve the efficacy of standard therapy in AML.9,55
Statins possess several mechanisms, such as apoptosis, antiproliferation,autophagy,to realize the anti-leukemia effect.Xia et al reported that lovastatin could induce apoptosis in AML.After conducting a series of add-back experiments, depressed geranylgeranylation of target proteins is identified as the predominant mechanism.56Through abrogating ERK MAP kinase pathway activation and affecting normal cell cycle,fluvastatin and atorvastatin could inhibit cell growth and enhance cytotoxicity in aggressive natural killer cell leukemia.57Lovastatin exposure increased the expression of leukocyte integrins CD11b and CD18 and decreased the level of BCL-2,inducing a pronounced differentiation response followed by extensive apoptosis in AML.58In rat basophilic leukemia(RBL-2H3)cells,simvastatin inhibited histamine release by suppressing the geranylgeranylation of Ras-like protein in the brain 27a(Rab27a) and interfering with the Rab27a-Doc2a (double C2 alpha)interaction,which influenced exocytosis in RBL-2H3 cells finally.59By reducing cholesterol levels,statins evoked autophagy in leukemic cells, as a consequence inactivated the main autophagy suppressor mTOR and its substrate ribosomal p70S6 kinase (p70S6K).60
However,leukemia cells are observed to display heterogenous responses to statins.For example,Wong et al demonstrated that AML cells had variant sensitivity to lovastatin, fluvastatin,atorvastatin,and cerivastatin.They found cerivastatin is at least 10 times more powerful than the other statins at inducing apoptosis.61Weide et al reported that simvastatin only exerted ab inhibitory effect on partial primary sorted CD34+ AML cells53and this heterogeneity attributes to the differences in Ras isoprenylation.52These findings indicate a subgroup of patients may need co-treatment.
5.2.FDPS and GGPS inhibitors
Deriving from the same biosynthetic pathway and reacting both primarily due to depletion of downstream products,GGPS inhibitor shares similar function with FDPS inhibitor.
Bisphosphonates function by targeting FDPS and are extensively used to treat osteoporosis and other complications of bone and calcium metabolism,62,63such as metastatic bone disease associated with multiple myeloma.64,65Their benefit on leukemia therapy also has come to the surface.
A third-generation bisphosphonate, zoledronic acid (ZOL),was reported to synergistically augment the antileukemic efficacy of imatinib, hydroxyurea (HU), cytarabine (Ara-C), and daunorubicin (DNR).66By inhibiting the prenylation of Ras and Ras-related proteins,ZOL accelerated the extent of imatinib to eradicate Ph+clone67and induce S-phase cell cycle arrest and apoptosis in both imatinib-sensitive and -resistant CML cells.68Besides, leukemias derived from T cells are characterized by osteolytic bone lesions and hypercalcemia.Bisphosphonates cannot only inhibit cell proliferation directly by virtue of inhibition of isoprenoid biosynthesis, but also relief from hypercalcemia due to their binding to calcium ions.69,70For example, Ishikawa et al proved bisphosphonate incadronate could prevent cell growth of adult T-cell leukaemia cells.70
Interestingly, Agabiti et al recently explored a new class of bisphosphonates including digeranyl bisphosphonate (DGBP),which targeted the subsequent enzyme of GGPS instead of FDPS.71DGBP likewise could inhibit lymphocytic leukemia cell proliferation and apoptosis mediated through caspases and MEK/ERK pathways,69and its anti-tumor effect was more potent than zoledronate.Apart from DGBP,Reilly et al tested five more isoprenoid bisphosphonates targeting GGPS while having little or no activity to FDPS,which could disturb the cell viability in the K562 CML cells.72Combinations of FDPS and GGPS inhibitors could synergistically inhibit growth and induce apoptosis in leukemia.73
5.3.Farnesyl-transferase inhibitors (FTIs) and Geranyl transferase inhibitors (GGTIs)
As mentioned before, prenylated proteins, such as the Ras family,are frequentlyactivatedin leukemia,as a consequence great interests are obtained with respect to the prenylation inhibitor.
Among FTIs, tipifarnib, lonafarnib, and BMS-214662 have been investigated in hematologic malignancies74and numerous clinical trials have been undertaken in this arena.31Tipifarnib,the most extensively investigated FTI,displays a cytotoxic effect on primary sorted CD34+AML cells, accomplished by an amplification effect in conjunction with simvastatin.53Phase I-II studies demonstrate that tipifarnib contributes to 8 to 9.5%hematological improvement in refractory/poor-risk AML.75In the Phase II study SWOG S0432,patients ≥70 years with AML respond to all tipifarnib regimens with 20%overall response rate and toxicities were acceptable.76In a phase II-III trial of juvenile myelomonocytic leukemia(JMML),administration of tipifarnib followed by hematopoietic stem cell transplant (HSCT) is safe and yield a 51% initial response rate.77However, a phase III study comparing tipifarnib with best supportive care suggested that tipifarnib showed no survival advantage for untreated AML≥70 years old.A two-gene expression assay (RASGRP1: APTX gene expression ratio)may assist in predicting AML patients who would benefit from tipifarnib.75
Comparatively,the quantity of reports on GGTIs in leukemia has a limitation.Xia et al found that the GGTI-298 could induce a similar apoptotic effect as lovastatin in AML.56In ATL,GGTI-298 could reduce cell viability and induce G(2)/M phase accumulation.78
Reserved geranylgeranylation of RAS represents an important mechanism of FTI resistance.To overcome this,co-treatment of FTIs and GGTIs causes an accumulation of unprocessed N-RAS and inactive N-RAS-RAF complexes, inducing significant growth inhibition (>70%) in myeloid cell lines.79Moreover,FTIs and GGTIs demonstrate the function to reduce graft-versushost disease (GVHD) significantly via effects on CD4 effector T cells, alleviating the complication of HSCT.80However, GGTIs have been disappointingly toxic.It is inevitable to develop nextgeneration FTIs and GGTIs.
6.FUTURE DIRECTIONS
Understanding the connections between metabolic pathways and leukemia is extremely crucial for developing new therapeutic approaches.As we discussed in this review, cholesterol metabolism has a very important impact on leukemia in multiple aspects, providing a strong rationale for exploring inhibitors of this key metabolic activity.There has been inspiring progress in applying cholesterol synthesis inhibitors in leukemia treatment.Several drugs,such as statins and tipifarnib,have been approved for clinical trials.More efforts are still required to realize the clinical translation of those drugs.
Advances in precision medicine have shifted the focus toward individual differences in leukemia cases.Leukemia is characterized by heterogeneity,consisting of a group of subtypes according to morphology, immunology, cytogenetics, and molecular features (MICM).Previous researches verified that leukemia subtypes respond diversely to a certain inhibitor, and a certain subtype of leukemia responds differently to inhibitors who have similar pharmacological activity.It is intriguing to explore whether leukemia could be subclassed based on characteristic profiles of cholesterol metabolism.Moreover, there is space for optimization in the administration regimens.The ultimate goal is to select the most appropriate inhibitor or drug combination to achieve the best efficacy with the lowest side effects.
Another question that needs to be addressed is developing new targets.Cholesterol metabolism is intricate, with numerous catalytic genes and metabolites involved,our knowledge of which is insufficient in the contrast.For example, current researches shine the light on the inhibitors targeting the de novo synthesis pathway, while those targeting cholesterol transportation in subcellular organelle and esterification are poorly studied in leukemia.Further metabolomic study may be valuable to comprehensively interpret cholesterol metabolism.
In summary,we can draw a conclusion that the deregulation of cholesterol homeostasis contributes to clonal leukemia expansion and chemo-insensitivity.It has a great significance to dissect the pathogenic mechanisms thoroughly and further explore affordable and safe drugs in leukemia.
杂志排行

血液科学的其它文章
- Successful ex vivo expansion of mouse hematopoietic stem cells
- Cell cycle regulation and hematologic malignancies
- Will immune therapy cure acute myeloid leukemia?
- Engineered human pluripotent stem cell-derived natural killer cells: the next frontier for cancer immunotherapy
- Hematopoietic stem cell metabolism and stemness
- Epigenetic regulation of hematopoietic stem cell homeostasis