C3: Consensus Cancer Driver Gene Caller
2019-10-22ChenYuZhuChiZhouYunQinChenAiZongShenZongMingGuoZhaoYiYangXiangYunYeShenQuJiaWeiQiLiu2
Chen-Yu Zhu, Chi Zhou, Yun-Qin Chen, Ai-Zong Shen,Zong-Ming Guo, Zhao-Yi Yang, Xiang-Yun Ye*,Shen Qu*, Jia Wei*, Qi Liu2,*,j
1 Department of Endocrinology & Metabolism, Shanghai Tenth People’s Hospital, Bioinformatics Department, School of Life Sciences and Technology, Tongji University, Shanghai 200092, China
2 Department of Ophthalmology, Ninghai First Hospital, Ninghai 315600, China
3 R&D Information, Innovation Center China, AstraZeneca, Shanghai 201203, China
4 Shanghai Chest Hospital, Shanghai Jiaotong University, Shanghai 200240, China
5 Department of Pharmacy, The First Affiliated Hospital of University of Science and Technology of China, Hefei 230036, China
KEYWORDS Somatic mutation;Cancer driver genes;Consensus;Data integration;Web server
Abstract Next-generation sequencing has allowed identification of millions of somatic mutations in human cancer cells.A key challenge in interpreting cancer genomes is to distinguish drivers of cancer development among available genetic mutations.To address this issue,we present the first webbased application, consensus cancer driver gene caller (C3), to identify the consensus driver genes using six different complementary strategies, i.e., frequency-based, machine learning-based, functional bias-based,clustering-based,statistics model-based,and network-based strategies.This application allows users to specify customized operations when calling driver genes, and provides solid statistical evaluations and interpretable visualizations on the integration results.C3 is implemented in Python and is freely available for public use at http://drivergene.rwebox.com/c3.
Introduction
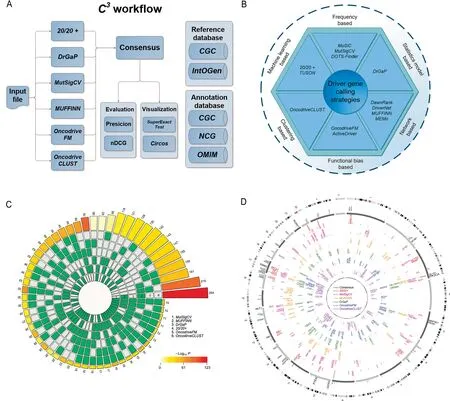
Figure 1 Guideline of C3 web server
The continued advancement of next-generation sequencing(NGS)technology has allowed for the sequencing of large sets of cancer samples for somatic mutation discovery [1,2]. However,one of the main challenges in interpreting the cancer genomes is to efficiently distinguish the driver mutations from the passenger mutations.Driver mutations are causally implicated in oncogenes and positively selected along the lineage of cancer development under the specific microenvironment conditions in vivo, whereas passenger mutations do not confer clonal growth advantages and are thus irrelevant to tumor development [3]. To address this issue, various methods have been proposed to identify driver genes based on distinctive assumptions and strategies [4-16]. Intuitively, all these driver gene identification strategies exhibit the biased signals of positive selection exploited by corresponding mechanisms at varied degrees. Several studies have been reported on benchmarking these methods with consensus cancer driver genes derived from individual model [8,17,18]. Collin et al. [8] proposed an evaluation framework to benchmark several existing models based on several measurements including precision, consistency, and mean log fold change (MLFC). Matan et al.[17] also benchmarked the available methods by using measurements such as precision and recall. Eduard et al. [18] classified four subtypes of driver gene calling methods at a subgene resolution. Denis et al. [19] provided the most comprehensive benchmarking of 21 driver gene prediction methods and proposed a Borda-based integration approach ConsensusDriver.
Despite these efforts,the available tools are often challenging for biologists or clinicians to carry out the related analysis directly,given the technical hurdles ranging from setting up the software to tuning parameters.A web-based user-friendly consensus driver gene prediction with intuitive visualization of the consensus mutation calling is needed.Here,we present the first web server-based consensus cancer driver gene caller(C3)platform to derive the consensus mutation calling results [4-17],using six state-of-the-arts and complementary prediction strategies. These include frequency-based (MutSigCV) [6],machine learning-based (20/20+) [8], functional bias-based(OncodriveFM) [10], clustering-based (OncodriveCLUST)[11], statistics model-based (DrGaP) [5], and network-based(MUFFINN) [7]. Various calling evaluation and visualization strategies are incorporated in C3as follows. (1) C3provides a solid evaluation of the consensus mutation calling results with Top-N-Precision and Top-N-nDCG [20]. (2) C3provides an efficient integration strategy to derive the consensus results by Robust Rank Aggregation (RRA) [21] and statistical model-based intersection visualization [22]. (3) Circos plots are presented in C3to visualize the consensus mutation calling results [22,23].
Method
General workflow of C3
C3accepts mutation annotation format (MAF) [24] file as input. The MAF file is annotated from variant calling format(VCF)[25]file,which can be acquired by using variant calling tool like Mutect on the NGS data.A schematic representation of the C3workflow is shown in Figure 1A. The selected programs, including 20/20+, MutSigCV, OncodriveFM, OncodriveCLUST, DrGaP, and MUFFINN (Figure 1A and B;File S1 Part 1), run in the Ubuntu sever 16.04 system. Then all preprocessed input mutation data are processed in C3to obtain candidate driver genes list for each strategy separately.We use SuperExactTest model to evaluate the statistical significance of the intersection of individual calling results using all the protein-coding gene as a whole background gene set. In addition, based on each discrepant driver gene list, a rank ensemble method,RobustRankAggreg,is used to obtain a consensus driver gene list. Four databases including the Cancer Gene Census (CGC) [26], Integrative Onco Genomics (IntOGen) [10], Network of Cancer Genes (NCG) [27], and Online Mendelian Inheritance in Man(OMIM)[28]are used to annotate the predicted driver genes.Two evaluation measurements,i.e., the Top-N-Precision and Top-N-nDCG, are applied to evaluate the calling performance.Finally,the KEGG[29]pathway and Gene Ontology analyses are also performed on the consensus driver genes for comprehensive annotations.
Performance measurement
Previously, Collin et al. proposed a novel measurement of mean log fold change between the observed and desired theoretical P values [8]. Matan et al. [17] and Eduard et al. [18]applied measurements of precision and recall.Denis et al.also applied precision, recall, and F1 score [19] (File S1 Part 1). In our study,we applied the Top-N-Precision(using CGC data as a reference driver gene set[26])and Top-N-nDCG(using IntOGen as a reference ranking driver gene set[30])to facilitate the quantitative comparison and evaluation,focusing on the top n performance of the ranking results.
Precision
We evaluated the precision performance among the results acquired by the previous strategies based on the top 100 genes with respect to CGC cancer database through Equation (1).The average precision can measure a general predicting ability of individual methods among the pan-cancer cohort samples.We calculate the precision scores for each of 27 cancer types,and the SUM (precision) represents the sum of respective precision score of 27 cancer types (Equation (2)).
Top-n-precision

nDCG
Meanwhile, normalized discounted cumulative gain (nDCG)was applied to measure the ranking quality of the results using the IntOGen as a reference cancer driver gene set.Weight of a reference gene


Figure 2 General framework of C3 web application

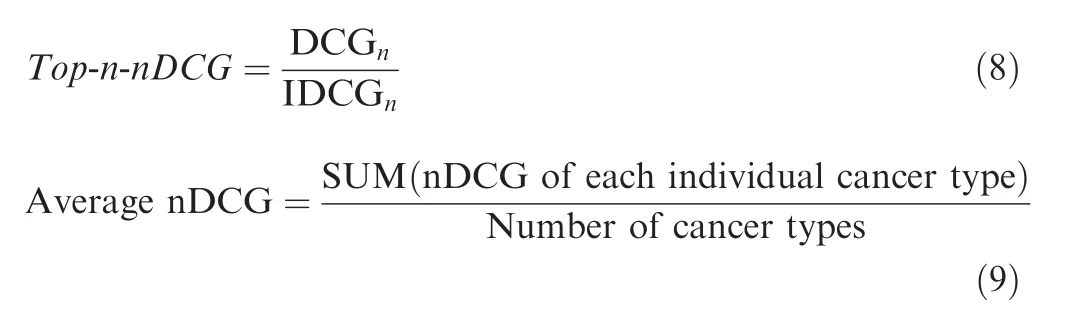
Here,n represents the number of top predicted genes;i represents the rank of predicted genes;CGnrepresents cumulative weight of top n predicted genes; DCGnrepresents CGnmultiplied by a discount factori(i >1); IDCGnrepresents a DCGnunder the ideal condition, that is, the rank of predicted genes is exactly the same as that in the reference dataset. Top-N-nDCG represents normalized DCGnand measures the ranking performance of predicted genes.
To obtain the Top-N-nDCG, firstly, we download IntOGen cancer driver gene set (URL: https://www.intogen.org/) [31]and assign a weight for each reference driver gene in IntOGen based on their proportion of driver mutation counts[30](Version 2014.12)calculated according to Equation(3).Specifically,the total number of cancer driver genes in IntOGen is 459.The weights of the predicted driver genes overlapping with the benchmark IntOGen dataset are calculated according to Equation (4). The weights of the predicted genes that are not available at the benchmark IntOGen dataset are set to 0. The Top-N-nDCG can be calculated through Equations(5)-(8)[20].
Rank aggregation
The RRA algorithm[21]is applied to obtain a consensus driver gene list, which aggregates the ranking driver genes predicted by individual tools. Comparing with the original RankAggreg algorithm[32],the RRA algorithm has three advantages:(1)it deals with incomplete rankings, which is common in practice,(2) it performs robustly with tolerance to the data noise, and(3) it is fast to be integrated for interactive data analysis.
Intersection visualization and evaluation with SuperExactTest and Circos
We applied SuperExactTest [22] and Circos [23] to organize our visualization results.The former is a scalable visualization tool to illustrate high-order relationships among multi sets beyond Venn diagrams [33]. It evaluates the overlap of each of tools and presents a circular plot illustrating all possible intersections with statistical methods. The latter visualizes the predicted driver gene sets intuitively(Figure 1C and D;File S1 Part 5).
Implementation
As Figure 2 shows,C3web application accepts MAF[24]file or a modified micro-MAF file(Table S1)as the input.After users select driver gene calling strategies and parameters,C3runs as the back-end Ubuntu 16.04 system (with python-2.7, R-3.3.4 and MATLAB Runtime 2014). When the job is successfullyfinished, users will be notified through email including a‘‘Request ID”. At the ‘‘Recent Request” page, users can preview and obtain candidate driver gene list by querying the‘‘Request ID”. The output is directly viewable on the website and is available to downloaded for further analyses. The data submitted by every user are kept private. If there are any questions, users can visit the ‘‘Help” page for a detailed guidance.

Table 1 Number of tested tumor samples and mutations

Figure 3 Comparison of cancer driver gene calling performance using Consensus and the six individual strategies on 27 cancer datasets
Detailed information of the test datasets
We test the stability of C3web application by selecting tumor datasets collected from The Cancer Genome Atlas(TCGA)[2]databases.Initially,the whole dataset includes 34 cancer types with 7724 samples and 729,235 mutations, curated from the published whole-exome sequencing or whole-genome sequencing studies which are also used by TUSON[9]and Collin study[8]. Since some tools (such as MutSigCV and DrGaP) need additional cohort mutation information, we removed 7 cancer types with 290 samples and 5164 mutations through data preprocessing.Finally,we curated 27 cancer types with 7434 samples and 724,071 mutations for the final analysis, which constitute the updated comprehensive test datasets finally for driver gene calling (Table 1 and File S1 Part 2).
Performance of C3
We benchmarked the performance of the consensus results comparing with each alternative. As shown in Figure 3, the integration results of C3application outperformed other methods evaluated with Top-N-Precision and Top-N-nDCG,revealing its superiority in driver genes prediction (File S1 Part 4).
C3also helps to identify reliable potential driver genes by SuperExactTest intersection between different driver gene calling strategies with reference to CGC and literature review.Detailed results are shown in Table S2 and Table S3.
In summary, although there exists a high discrepancy among different driver gene identification strategies, the intersection by individual strategies not only identifies the most reliable driver genes, but also helps to find potential novel driver genes that are not well-characterized.
Future developments
Currently C3has some limitations and warrants future updates. (1) C3is currently deployed on the Ali Cloud server,which requires a lot of memory and space to process the data.Any variant file exceeding 40,000 records may fail when running DrGaP. Since the Random Forest Model 20/20+occupies too much CPU resources, it also takes a long time(>3 h for sample of 50,000 mutations with 8 cores of Intel Xeon E5-2643 3.3 GHz) to run a whole pipeline of C3.Future optimizations are required to accelerate C3.(2)Current version of C3only supports the GRCH37 reference genome,and a new version of the reference genome such as GRCH38 will be added in the next version. (3) One potential application of C3is to identify the target driver genes for drug discovery.However, the computationally predicted drivers should not be over-interpreted without additional experimental evidence.
Availability
C3 is freely available for public use at http://drivergene.rwebox.com/c3.
Authors’ contributions
QL, JW, XY, and SQ conceived the project. CYZ, CZ, YC,and ZG designed the platform. CYZ, AS, and ZY analyzed the data. QL, YC, CZ, and CYZ wrote the manuscript. All authors read and approved the final manuscript.
Competing interests
The authors declare that they have no competing interests.
Acknowledgments
This work was supported by the National Major Research and Innovation Program of China (Grant Nos. 2017YFC0908500 and 2016YFC1303205),National Natural Science Foundation of China (Grant No. 61572361), Shanghai Rising-Star Program (Grant No. 16QA1403900), Shanghai Natural Science Foundation Program (Grant No. 17ZR1449400), and Fundamental Research Funds for the Central Universities (Grant No. 1501219106), China.
Supplementary material
Supplementary data to this article can be found online at https://doi.org/10.1016/j.gpb.2018.10.004.
杂志排行

Genomics,Proteomics & Bioinformatics的其它文章
- Diversified Application of Barcoded PLATO(PLATO-BC) Platform for Identification of Protein Interactions
- gFACs: Gene Filtering, Analysis, and Conversion to Unify Genome Annotations Across Alignment and Gene Prediction Frameworks
- H3K27me3 Signal in the Cis Regulatory Elements Reveals the Differentiation Potential of Progenitors During Drosophila Neuroglial Development
- Characterization of Distinct T Cell Receptor Repertoires in Tumor and Distant Non-tumor Tissues from Lung Cancer Patients
- Warburg Effects in Cancer and Normal Proliferating Cells: Two Tales of the Same Name
- Proteomics Analysis of Lipid Droplets from the Oleaginous Alga Chromochloris zofingiensis Reveals Novel Proteins for Lipid Metabolism