哒嗪分子的叠氮基衍生物作为含能分子的理论研究
2019-10-21李璐琳
李璐琳

摘 要:本文通过用叠氮基逐次取代哒嗪分子上的氢原子的方式设计了一系列叠氮基哒嗪衍生物分子。通过设计均裂反应,计算了这些分子的解离能;通过设计等键反应,计算了这些分子的生成焓。通过这些参数的计算,考察了标题分子的动力学和热力学稳定性。为了评估标题分子作为含能分子的性能,通过Kamlet-Jacobs方程计算了标题分子的爆热、爆压、爆速和分子密度。计算结果表明,综合考虑结构稳定性和爆轰性能,三个分子是潜在的高能量密度分子。
关键词:密度泛函理论;量子化学计算;含能材料
中图分类号:TQ560.1文献标识码:A文章编号:1003-5168(2019)16-0127-03
Abstract: In this paper, a series of azidopyridazines derivatives had been designed by replacing hydrogen atoms on pyridazines with azido groups step by step. The dissociation energy of these molecules was calculated by design homogeneous reaction, and the enthalpy of formation of these molecules was calculated by designing equal bond reaction. Through the calculation of these parameters, the kinetics and thermodynamic stability of the title molecule were investigated. In order to evaluate the performance of the title molecule as an energetic molecule, the detonation heat, explosion pressure, detonation velocity and molecular density of the title molecule were calculated by Kamlet-Jacobs equation. The results show that the threemolecules are potential high energy density molecules considering structural stability and detonation performance.
Keywords: density functional theory;quantum chemistry;energetic materials
高能量密度分子因在工業和军事上具有巨大的应用价值而受到广大科学家的广泛关注。目前,实验室中已经获得大了大量的高能量密度分子,如TATB、RDX和CL-20等[1-3]。从这些高能量密度分子来看,高的氮含量是其共同特征。因此,向高氮母体结构中引入高氮含能基团是设计一个高能量密度分子的有效手段。
哒嗪分子,是一个具有两个氮原子的六圆环结构,其氮含量达53%。同时,在六圆环上具有共轭的π键,为分子带来了额外的芳香稳定性。显然,哒嗪是一个较好的含能分子的母体结构,既有高的氮含量,又兼顾了稳定性[4,5]。
因此,本文采取向哒嗪分子中逐次用叠氮基取代氢原子的方法,设计了一系列高氮分子,考察了这类分子的热力学、动力学稳定性及爆轰性能,希望为这一类分子未来的实验室研究提供理论支持。
1 计算方法
我们设计的所有分子的结构优化都使用Gaussian09程序包完成,使用的理论方法为B3LYP/6-311G(d,p),进一步通过频率计算确定了这些结构在势能面是能量极小点。生成焓的计算通过设计等键反应来实现,反应方程式为:
C4N2+3nH4-n+nCH4→nCH3N3+C4N2H4 (1)
(2)
在炸药的爆轰性能中,爆速(D)和爆压(P)是衡量含能材料爆轰性能的两个重要指标。对于C、H、N和O组成的含能体系,其D和P值可用K-J方程估算得到。
2 结果与讨论
2.1 结构与稳定性
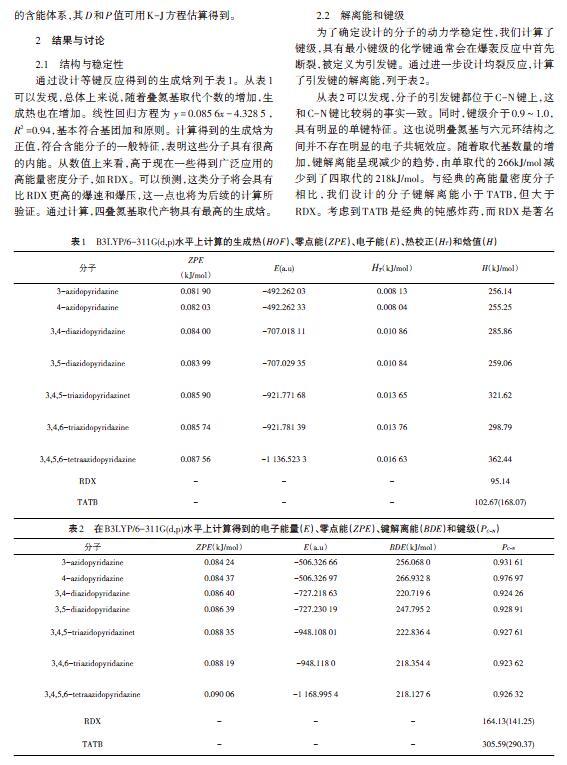
通过设计等键反应得到的生成焓列于表1。从表1可以发现,总体上来说,随着叠氮基取代个数的增加,生成热也在增加。线性回归方程为[y=0.085 6x-4.328 5],[R2]=0.94,基本符合基团加和原则。计算得到的生成焓为正值,符合含能分子的一般特征,表明这些分子具有很高的内能。从数值上来看,高于现在一些得到广泛应用的高能量密度分子,如RDX。可以预测,这类分子将会具有比RDX更高的爆速和爆压,这一点也将为后续的计算所验证。通过计算,四叠氮基取代产物具有最高的生成焓。
2.2 解离能和键级
为了确定设计的分子的动力学稳定性,我们计算了键级,具有最小键级的化学键通常会在爆轰反应中首先断裂,被定义为引发键。通过进一步设计均裂反应,计算了引发键的解离能,列于表2。
从表2可以发现,分子的引发键都位于C-N键上,这和C-N键比较弱的事实一致。同时,键级介于0.9~1.0,具有明显的单键特征。这也说明叠氮基与六元环结构之间并不存在明显的电子共轭效应。随着取代基数量的增加,键解离能呈现减少的趋势,由单取代的266kJ/mol减少到了四取代的218kJ/mol。与经典的高能量密度分子相比,我们设计的分子键解离能小于TATB,但大于RDX。考虑到TATB是经典的钝感炸药,而RDX是著名的猛炸药,本文设计的分子的动力学稳定性普遍是比较优秀的。
2.3 爆轟性能
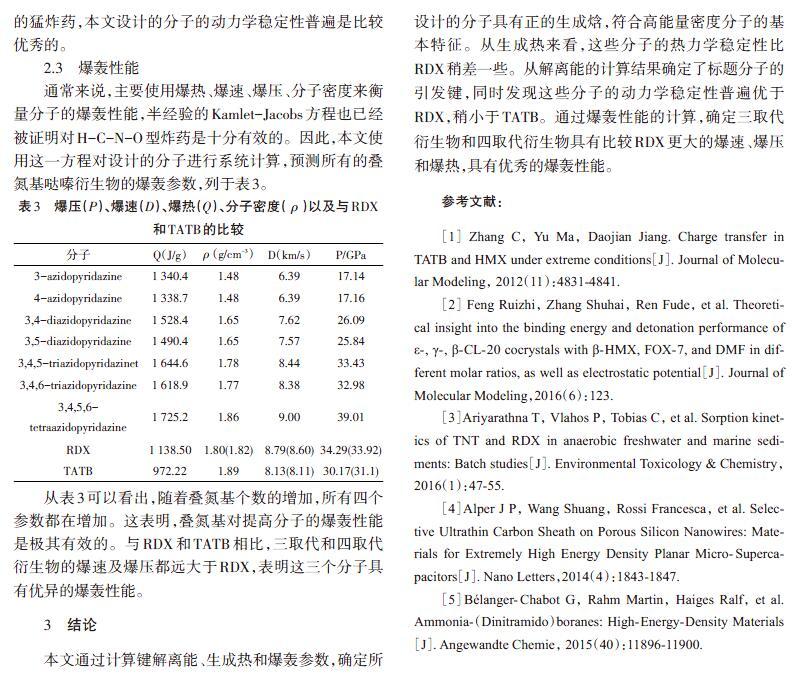
通常来说,主要使用爆热、爆速、爆压、分子密度来衡量分子的爆轰性能,半经验的Kamlet-Jacobs方程也已经被证明对H-C-N-O型炸药是十分有效的。因此,本文使用这一方程对设计的分子进行系统计算,预测所有的叠氮基哒嗪衍生物的爆轰参数,列于表3。
从表3可以看出,随着叠氮基个数的增加,所有四个参数都在增加。这表明,叠氮基对提高分子的爆轰性能是极其有效的。与RDX和TATB相比,三取代和四取代衍生物的爆速及爆压都远大于RDX,表明这三个分子具有优异的爆轰性能。
3 结论
本文通过计算键解离能、生成热和爆轰参数,确定所设计的分子具有正的生成焓,符合高能量密度分子的基本特征。从生成热来看,这些分子的热力学稳定性比RDX稍差一些。从解离能的计算结果确定了标题分子的引发键,同时发现这些分子的动力学稳定性普遍优于RDX,稍小于TATB。通过爆轰性能的计算,确定三取代衍生物和四取代衍生物具有比较RDX更大的爆速、爆压和爆热,具有优秀的爆轰性能。
参考文献:
[1] Zhang C, Yu Ma, Daojian Jiang. Charge transfer in TATB and HMX under extreme conditions[J]. Journal of Molecular Modeling, 2012(11):4831-4841.
[2] Feng Ruizhi, Zhang Shuhai, Ren Fude, et al. Theoretical insight into the binding energy and detonation performance of ε-, γ-, β-CL-20 cocrystals with β-HMX, FOX-7, and DMF in different molar ratios, as well as electrostatic potential[J]. Journal of Molecular Modeling,2016(6):123.
[3]Ariyarathna T, Vlahos P, Tobias C, et al. Sorption kinetics of TNT and RDX in anaerobic freshwater and marine sediments: Batch studies[J]. Environmental Toxicology & Chemistry,2016(1):47-55.
[4]Alper J P, Wang Shuang, Rossi Francesca, et al. Selective Ultrathin Carbon Sheath on Porous Silicon Nanowires: Materials for Extremely High Energy Density Planar Micro-Supercapacitors[J]. Nano Letters,2014(4):1843-1847.
[5]Bélanger-Chabot G, Rahm Martin, Haiges Ralf, et al. Ammonia-(Dinitramido)boranes: High-Energy-Density Materials[J]. Angewandte Chemie, 2015(40):11896-11900.
