头孢洛扎的7-位侧链噻二唑衍生物TATD的制备方法
2019-10-10周军荣张佳江东杨彪
周军荣 张佳 江东 杨彪
(浙江东邦药业有限公司,临海 317016)
头孢洛扎的7-位侧链噻二唑衍生物的化学名为:(Z)-2 - [(5-氨基-[1,2,4]噻二唑-3-基)-羧基-亚甲基氨基氧基]-2-甲基-丙酸叔丁酯[1],简称TATD,其化学结构如图1所示。

图1 TATD的结构式Fig.1 Structure of TATD
TATD作为合成新型抗生素头孢洛扎的重要中间体,具有巨大的社会经济效益。头孢洛扎属于第五代头孢菌素,具有抗菌谱广、高效的特点,对多种革兰阴性菌表现出极佳的抗菌活性,与头孢西丁、头孢曲松等相类似。头孢洛扎与其他头孢菌素的不同在于大大地增强了对于铜绿假单胞菌的活性,甚至包括碳青霉烯和哌拉西林/三唑巴坦(β-内酰胺酶抑制剂)的耐药株,且对革兰阳性的链球菌非常有效[2-3]。但是头孢洛扎对产超广谱β-内酰胺酶(ESBLs)的细菌基本无效,因此将它与三唑巴坦组成复方静脉注射剂,增加对产ESBLs菌株的活性,对厌氧菌也具有活性。于2014年,头孢洛扎/三唑巴坦在美国经FDA批准上市,商品名为Zerbaxa®,是第四个被FDA批准并指定为合格的感染疾病产品(QIDP)新抗菌药产品。该药物的半衰期长、不良反应发生率低,主要用于治疗患有复杂性腹腔内感染(cIAI)及复杂性尿路感染(cUTI)的成年患者[4],满足由更广泛的耐药革兰阴性菌所带来的治疗需求,前景可期。
目前有相关专利及文献提及到TATD的使用,但是并未公布报道它的制备方法。当然在头孢菌素侧链的合成方法中,也具有类似结构的合成。比如国内专利[5]公开了类似化合物A(头孢他啶侧链酸活性酯)的合成路线,如图2所示的路线;再如国内专利及文献[6-7]公开了类似化合物B(头孢唑兰侧链酸)的合成路线,具体见图3所示;还有国外文献[8-10]报道了类似化合物C的合成方法,如图4所示的路线。
由于上述的类似化合物A~C的结构与TATD不完全相同,有关TATD的合成方法尚未见公开报道。因此,开发一种头孢洛扎的7-位侧链噻二唑衍生物TATD的制备方法,具有巨大的社会意义及经济效益。
参考上述文献及专利提及到合成路线,设计出了一条合成TATD的工艺路线,具体路线如图5所示。该工艺路线首先以2-肟氰基乙酸乙酯与2-溴代异丁酸叔丁酯为起始原料,进行缩合反应得到中间体1;中间体1与氨水反应得到酰胺化合物中间体2;然后再依次进行脱水反应、脒基化反应与水解反应得到中间体3,其中3个反应使用“一锅法”进行;最后中间体3与硫氰酸铵进行合环反应得到TATD。

图2 类似化合物A的合成路线Fig.2 Synthetic route of similar compound A

图3 类似化合物B的合成路线Fig.3 Synthetic route of similar compound B

图4 类似化合物C的合成路线Fig.4 Synthetic route of similar compound C

图5 TATD的合成路线Fig.5 Synthetic route of TATD
1 实验部分
1.1 仪器与药品
实验仪器Varian 400-MR型核磁共振波普仪,梅特勒托利多FE28-Standard实验室台式pH计酸度计,Agilent 1200/1260高效液相色谱仪,AR1140电子天平,ALPHA傅里叶变换红外光谱仪等。
主要原料2-肟氰基乙酸乙酯(上海易恩化学技术有限公司),2-溴代异丁酸叔丁酯(上海易恩化学技术有限公司),其它溶剂与试剂均来源于上海国药集团。
1.2 合成过程
1.2.1 中间体1的合成
常温下,向反应瓶中加入140mL DMF、2-肟氰基乙酸乙酯20g(0.14mol, 1eq)和三乙胺25.5g(0.25mol, 1.80eq),搅拌均匀。控温20~25℃,滴加2-溴代异丁酸叔丁酯43.73g(0.196mol, 1.4eq);滴加完毕,升温至30~35℃,进行缩合反应3h(展开剂为石油醚:乙酸乙酯=2:1,V/V,TLC跟踪监测反应)。反应结束后,在常温下加入180mL甲基叔丁基醚与100mL水,用适量的浓盐酸调节反应液体系的pH值至1.5~2.0。之后静置、分液,将水层弃掉,收集有机层待用。常温下,往有机层料液中加入120mL水,搅拌洗涤5min。之后静置、分液,将水层弃掉,收集有机层待用。控温35℃以下,将收集的有机层进行减压浓缩至油状物,即得到36.8g中间体1,摩尔收率为92.5%,HPLC检测(色谱柱:Develosil ODS-P-5 150mm×4.6mm, 5μm;检测波长:254nm;柱温:25℃)纯度为99.0%;ESIMS:m/z284.14[M+H]+。
1.2.2 中间体2的合成
常温下,向反应瓶中加入210mL甲醇与30g(0.105mol, 1eq)中间体1,然后开始滴加33g 20%(0.39mol, 3.71eq)的氨水。滴加完毕,将反应体系升温至35~40℃,保温反应2h(展开剂为石油醚:乙酸乙酯=2:1,V/V,TLC跟踪监测反应)。反应结束后,向料液中加入160mL水,用适量浓盐酸调节pH值为2.5~3.0,将料液控温在40℃以下,进行减压浓缩,浓缩至一定体积,将料液降温至5~10℃,搅拌析晶2h。析晶结束后,将物料进行过滤,将湿品置于真空干燥箱中,控温35~40℃,干燥10h左右。最后得到干品24.5g中间体2,水分<0.5%,摩尔收率为91.4%,HPLC检测(色谱柱:Develosil ODS-P-5, 150mm×4.6mm, 5μm;检测波长:254nm;柱温:25℃),纯度为98.8%;ESI-MS:m/z255.12[M+H]+。
1.2.3 中间体3的合成
常温下,向反应瓶中加入400mL甲基叔丁基醚与40g(0.16mol, 1eq)中间体2,搅拌溶解。控温15℃以下,加入77g(0.37mol, 2.31eq)五氯化磷,加完之后,再滴加40g(0.51mol, 3.19eq)吡啶。滴加完毕,并控温20~25℃,进行脱水反应3h(展开剂为石油醚:乙酸乙酯=2:1,V/V,TLC跟踪监测反应)。反应结束后,常温下,往反应液中加入260mL水,搅拌洗涤5min。之后静置、分液,将水层弃掉,收集有机相待用。控温25℃以下,将得到的有机层进行减压浓缩得到油状物过渡态1。在常温下,将450mL甲醇加至上述油状物过渡态1中,搅拌溶解。控温在20~30℃,加入30g(0.56mol, 3.5eq)甲醇钠,搅拌反应5h。之后加入35g(0.65mol, 4.06eq)氯化铵,并升温至50~55℃,保温反应8h(展开剂为石油醚:乙酸乙酯=2:1,V/V,TLC跟踪监测反应)。反应结束后,在常温下,滴加入50%氢氧化钠水溶液105g(1.31mol, 8eq),控温50~55℃,进行水解反应2h(展开剂为二氯甲烷:甲醇=6:1,V/V,TLC跟踪监测反应)。应结束后,常温下,加入适量浓盐酸调节体系的pH值为3.0~3.5。控温30℃以下,将料液进行减压浓缩至一定体积。浓缩完毕,再降温至5~10℃,搅拌析晶2h。析晶结束后,将物料进行过滤,将湿品置于真空干燥箱中,控温40~45℃,干燥10h左右,得到31.5g中间体3,水分<0.5%,摩尔收率为73.5%,HPLC检测(色谱柱:Develosil ODS-P-5 150mm×4.6mm, 5μm;检测波长:254nm;柱温:25℃)纯度为99.1%;ESI-MS:m/z273.13[M+H]+。
1.2.4 TATD的合成
常温下,向干净的反应瓶中加入300mL甲醇、25g(0.09mol, 1eq)中间体3、23g(0.30mol, 3.33eq)硫氰酸铵与23.5g(0.23mol, 2.56eq)三乙胺,搅拌溶解。然后控温5℃以下,缓慢滴加5.04g(0.03mol, 0.35eq)溴。滴加完毕,将反应体系的温度升至30~35℃,进行合环反应4h(展开剂为二氯甲烷:甲醇=6:1,V/V,TLC跟踪监测反应)。反应结束后,控温在35℃以下,将反应液进行减压浓缩至150mL。再控温20~30℃,用适量的10%稀盐酸调节pH值为1.5~2.0。调节完毕,常温下继续搅拌析晶2h。析晶结束后,将物料进行过滤,将湿品置于真空干燥箱中,控温35~40℃,干燥10h左右,得到25.3g TATD,水分<0.5%,摩尔收率为85.0%,HPLC检测(色谱柱:Develosil ODS-P-5 150mm×4.6mm, 5μm;检测波长:254nm;柱温:25℃)纯度为99.3%;1H NMR(400MHz,CDCl3) δ(ppm):4.86(s, 2H, NH2),3.28(s, 6H, CH3),1.47(d, 9H, CH3);ESI-MS:m/z330.10[M+H]+;IR(KBr)3403、3308、3211、2980、2939、1758、1716、1634、1538、1472、1418、1295、1241、1149、995、851和598cm-1。
2 分析讨论
2.1 中间体1的合成
该步骤是以2-肟氰基乙酸乙酯与2-溴代异丁酸叔丁酯为起始原料,进行缩合反应。保持其他实验条件与“1.2.1”项的中间体1的合成步骤相同,改变2-溴代异丁酸叔丁酯的当量,产物中间体1纯度及摩尔收率见表1。由表1可以看出,当2-溴代异丁酸叔丁酯与2-肟氰基乙酸乙酯的摩尔比在1.0~1.4:1.0的改变过程中,随着2-溴代异丁酸叔丁酯用量的增加,产品的纯度及收率也基本随着增加。但当摩尔比超过1.4:1.0之后,目标化合物的收率变化不明显,而纯度有所下降,原因可能为未反应完的2-溴代异丁酸叔丁酯残留在产品中,导致纯度降低。考虑到物料成本及产品的收率与纯度,因此2-溴代异丁酸叔丁酯与2-肟氰基乙酸乙酯的摩尔比在1.4:1.0为最佳的物料比。
该反应为脱溴化氢反应,选择一种合适的缚酸剂具有较大的意义。研究该反应时,考察过使用不同的缚酸剂(有机碱包括吡啶、三乙胺、二乙胺、二异丙胺、哌啶及吡咯)对反应结果的影响,前提是其他实验条件与“1.2.1”项中中间体1的合成步骤相同,具体见表2。由表2结果显示,使用三乙胺作为缚酸剂时,反应得到的产物的收率及纯度经综合考虑是最佳的。
反应结束后处理时,用浓盐酸进行调节pH为1.5~2.0,主要为了去除未反应的三乙胺,避免三乙胺残留在产品中,以提高产物中间体1的纯度。
2.2 中间体2的合成
该步骤是以中间体1与氨水发生酯氨解反应得到中间体2,该反应比较容易进行。关键是选择一种合适的溶剂,对该反应有促进作用。选择醇类溶剂既能溶解原料中间体1,也能与氨水互溶,使反应体系处于均相体系,能够使反应充分进行。尝试过使用甲醇、乙醇与乙二醇,结果发现使用甲醇时,其收率及纯度是最佳的,具体见表3。

表1 2-溴代异丁酸叔丁酯的用量对合成中间体1的影响Tab.1 Effect of the amount of t-Butyl 2-bromo isobutyrate on the synthesis of intermediate 1
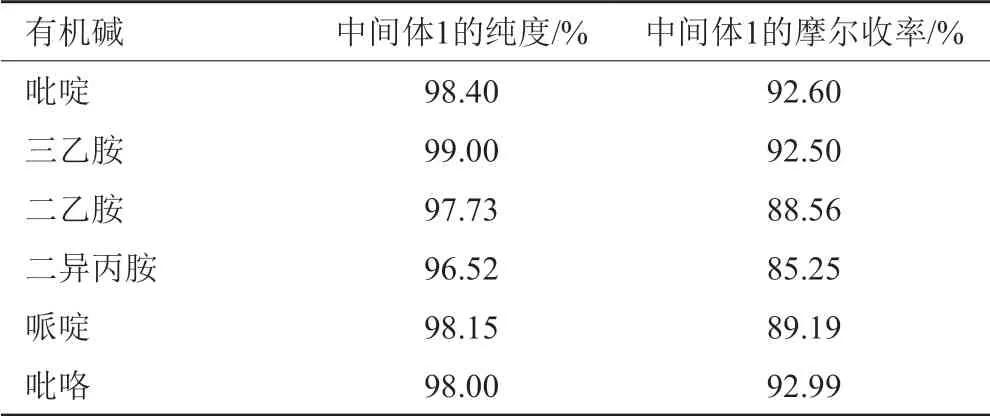
表2 不同有机碱对合成中间体1的影响Tab.2 Effect of different organic bases on the synthesis of intermediate 1

表3 不同溶剂对合成中间体2的影响Tab.3 Effect of different solvents on the synthesis of intermediate 2
2.3 中间体3的合成
该步骤是将脱水反应、脒基化反应及水解反应进行“一锅法”反应,减少出料损失,避免了浓缩和长时间的存放周期,能够有效避免羧酸酯分解形成副产物,提高产物的收率和纯度。脱水反应选择脱水剂时,考察过五氯化磷与三氯氧磷,虽然两者的反应效果差不多,但是三氯氧磷属于剧毒品,对环境污染较为严重,所以综合考虑选择五氯化磷。
脒基化反应是一个缓慢的过程,因此脒基化反应时间是一个关键点。考察反应时间时,前提是其他实验条件与“1.2.3”项中间体3的合成步骤相同。其反应时间较短时,原料残留较多,导致收率低,纯度差;当反应时间过久,脱水反应生成的过渡态1化合物中的两个氰基结构均有可能被脒基化,导致副产物过大,具体见表4。由表4数据得知,所以脒基化最佳反应时间为8h,其产品的收率及纯度均较高。
水解反应时,考察研究过氢氧化钾或氢氧化钠的水溶液,实验发现两者的反应效果差不多,但是氢氧化钠的价格相对较低廉,所以综合考虑选择了氢氧化钠。其碱的用量对水解反应来说是个关键,探索碱的用量对产品的纯度及收率的影响,其他实验条件与“1.2.3”项中间体3的合成步骤相同,实验数据具体见表5。由表5结果显示,当氢氧化钠的用量为4eq时,发现原料残留较大,导致收率及纯度较差;当氢氧化钠的用量为5~10eq时,反应效果较好,纯度及收率均较高;当氢氧化钠的用量为11eq时,杂质较大,可能原因是碱性过强,导致化合物中的异丁酸叔丁酯结构水解生成副产物,得到产品纯度较差。综合考虑物料成本及产品的收率与纯度,氢氧化钠与中间体2的最佳摩尔比为8.0:1.0。
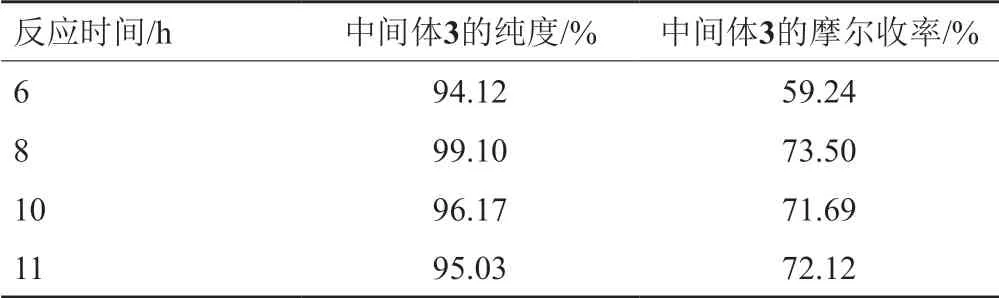
表4 不同反应时间对合成中间体3的影响Tab.4 Effect of different reaction time on the synthesis of intermediate 3

表5 氢氧化钠的用量对合成中间体3的影响Tab.5 Effect of the amount of NaOH on the synthesis of intermediate 3
2.4 TATD的合成
该步骤将中间体3与硫氰酸铵进行经典的噻二唑合环反应,并加入溴促进反应,因此探索溴的用量对于反应结果的影响是有意义的,前提是其他实验条件与“1.2.4”项TATD的合成步骤相同,实验数据具体见表6。由表6的数据看出,当不加溴时,反应进行的很慢,得到产品的收率及纯度均很低;随着溴的加入量增加,产品的收率先增加后基本不变,产品的纯度也是先提高后变化不明显。综合考虑,溴与中间体3的最佳摩尔比为0.35:1.0。

表6 溴的用量对合成TATD的影响Tab.6 Effect of the amount of Br2 on the synthesis of TATD
3 结论
本工艺是以2-肟氰基乙酸乙酯与2-溴代异丁酸叔丁酯为起始原料,进行缩合反应得到中间体1;中间体1与氨水反应得到酰胺化合物中间体2;然后将中间体2脱水反应、脒基化反应与水解反应使用“一锅法”进行反应得到中间体3;最后,将中间体3与硫氰酸铵进行噻二唑合环反应得到产品TATD。该工艺得到的TATD,不仅纯度及收率较高,而且所用原料易得,适合工业化生产。因此,开发出该工艺路线具有一定的经济效益与社会意义。
