Fe,Co,Ni掺杂GaSb的电子结构和光学性质*
2019-10-09潘凤春林雪玲曹志杰李小伏
潘凤春 林雪玲 曹志杰 李小伏
(宁夏大学物理与电子电气工程学院,银川 750021)
1 引 言
将高温热辐射体的能量通过半导体p-n结直接转换成电能的热光伏技术理念诞生于20世纪60年代.热光伏技术由于具有可广泛使用热源和能量输出密度高等独特的性能使其在尖端科研领域和军事上有很大的应用价值.热光伏电池是热光伏技术系统的核心部件,由于热辐射器辐射的光子能量主要处于红外光谱范围,因此对热光伏电池材料的禁带宽度要求相对较窄.最早的热光伏电池的材料是用禁带宽度为1.12 eV的Si材料制作的,不能很好地和热辐射器光谱相匹配,很多研究者用Yb2O3材料制作选择性辐射器和Si光伏电池配合制作的热光伏系统很好地解决了这个问题[1,2].Ge是一种禁带宽度只有0.66 eV的半导体材料,采用Er2O3制作选择辐射器可以较好地和Ge光伏电池相匹配[3,4].虽然Si,Ge光伏电池制作成本低廉,但由于光电转换效率不高,输出电压较低,使得热光伏系统没有得到重视.热光伏系统重新引起人们的极大关注得益于高效低禁带宽度的IIIV族半导体的出现.III-V族半导体大部分为直接带隙半导体,具有电子迁移率高、空穴迁移率高、缺陷密度低、性能稳定和易于生长等优点,在高频器件、半导体激光器、红外探测器和场效应晶体管等方面有广泛的应用[5−14].在III-V族化合物半导体中,GaSb的禁带宽度(0.812—0.813 eV)几乎为GaAs材料的一半,可以与多种辐射体的光谱相配合,且晶格常数为a=b=c=0.6095 nm,能与很多Sb化物材料、Sb化物二元及多元化合物的晶格常数相匹配[15],因此以GaSb材料为基础的热光伏电池得到了广泛的研究: Khvostikvo等[16]利用液相外延(liquid phase epitaxy,LPE)方法制备了热电转换效率为6%的GaAs/GaSb热光伏电池;Vlasov等[17]利用LPE和Zn扩散的方法制作了电池效率为8%的GaSb电池; Wang等[18]通过建立GaSb吸收系数的解析模型,给出了该电池短路电流、开路电压和填充因子随温度变化的曲线;Kim等[19]利用晶片规模加工技术在半绝缘GaAs衬底上制作了单片集成的GaSb电池阵列.为了使GaSb基热光伏电池更好地与温度较低的辐射器相匹配,更充分地利用红外区光谱以提高其转换效率,基于GaSb相关的三元、四元III-V族化合物被发现并用来制作热光伏电池,这类材料主要包括InGaSb,AlGaAsSb,InGaAsSb和InGaAsP等[20−28].多元GaSb相关化合物禁带宽度最低达到了0.53 eV,有效拓展了红外光谱的响应范围,光电转换效率也提高了很多,而且其表面附近的高内建电场提高了开路电压,但是较大的表面复合速度限制了该类材料的某些物理特性.目前用于热光伏电池材料的GaSb及其多元化合物仍然存在禁带宽度较大,不能很好地和热辐射器相匹配以及实际转换效率不高等问题,因此如何进一步拓展GaSb材料的红外光谱吸收范围成为研究的热点.研究表明[29−31],通过掺杂某些特定元素可以有效降低被掺杂半导体的禁带宽度和对半导体改性,基于此,本文采用基于密度泛函理论框架下的第一性原理计算方法研究了Fe,Co,Ni掺杂GaSb晶体的结构、电子结构和光学性质,期望我们的研究可扩展GaSb基半导体在红外热光伏电池、红外光探测器和红外半导体激光器等领域的应用.
2 研究方法与模型的构建
本文采用基于密度泛函理论的CASTEP[32,33]软件进行研究.将一个包含8个原子的GaSb原胞分别沿x,y,z三个方向进行2×2×2扩展,得到包含64个原子的超晶胞作为计算体系.电子体系波函数采用平面波波函数展开,平面波截断能为330 eV,体系电荷密度和总能量在布里渊区进行积分计算,采用Monkhorst-Pack[34,35]方案选取K空间网格点,布里渊区K网格点选取为3×3×3,基态能量采用Pulay密度混合法,自洽精度为5.0×10−7eV/atom,选取的赝势为超软赝势(ultra soft),交换关联泛函采用效率高且能正确反映固体电子密度及晶体结构特性的LDA-CAPZ泛函来处理.采用先进行几何结构优化再进行单点能量和光学性质的计算步骤进行.为了修正LDA/GGA交换关联泛函极大低估GaSb禁带宽度这一问题,Ga-3d电子和Sb-5p电子之间的关联能采用一个和轨道占据以及自旋相关的有效库仑作用能,即采用LDA+U方案将费米面附近连续分布的d能级和p能级分开从而得到正确的基态性质.本文采用作者以前计算过的数值:UGa-3d=2.5eV ,USb-5p=2.6eV[36].
包含64个原子的超晶胞结构如图1所示,图中大球表示Ga原子,小球表示Sb原子,为了描述方便,图中数字表示超晶胞中被掺杂元素替代的Ga原子位置,当用一个X替代图1中的3号原子,得到了包含一个X@Ga缺陷的超晶胞.当用一个X替代图1中的一个小球原子,便得到包含一个替代Sb缺陷(X@Sb)超晶胞.可以看出共有Fe@Ga,Co@Ga,Ni@Ga,Fe@Sb,Co@Sb和Ni@Sb六种基本的缺陷类型,这六种缺陷可以统一表示为X@Y,其中Y=Ga,Sb.本文所涉及的5种原子的价电子组态分别为Ga-3d104s24p1,Sb-4d105s25p3,Fe-3d64s2,Co-3d74s2,Ni-3d84s2.
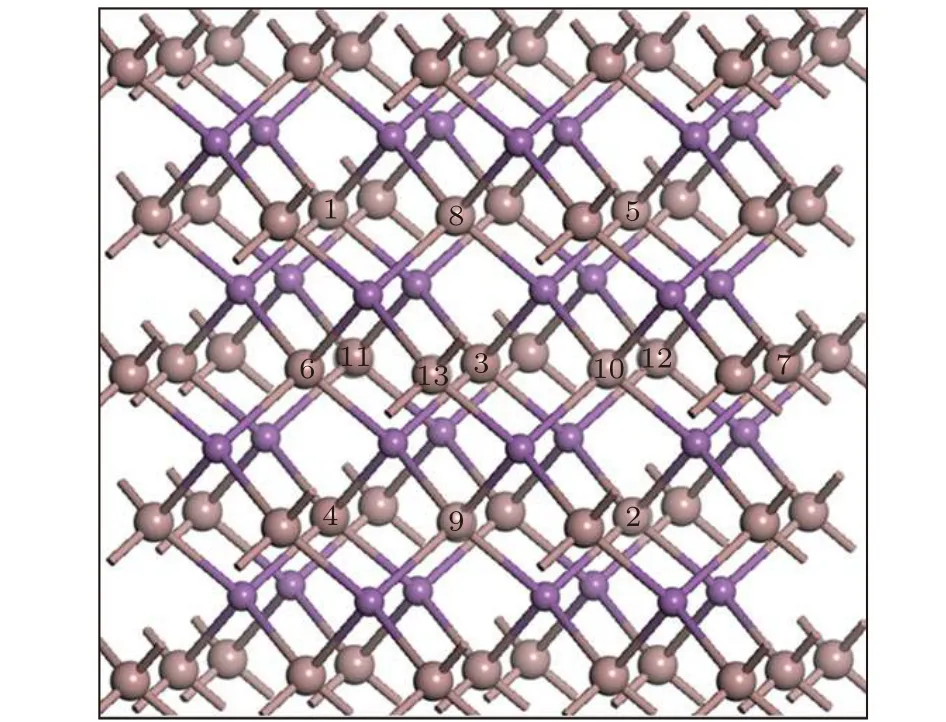
图1 GaSb超晶胞结构Fig.1.Structure of GaSb supercell.
3 计算结果和分析
3.1 包含X@Y缺陷体系的光学吸收谱
图2给出了包含一个X@Ga缺陷和一个X@Sb缺陷的GaSb超晶胞的光学吸收谱.图2(a)所示为包含一个X@Ga缺陷GaSb体系的光学吸收谱,可以看出,X掺杂的GaSb (X-GaSb)体系光学吸收谱相比于未掺杂的GaSb体系(黑色实线),其光学吸收谱的吸收边落在了远红外区,即发生了红移现象.特别地,Ni掺杂的GaSb (Ni-GaSb)体系(蓝色虚线)的光学吸收谱相比于Fe,Co掺杂的体系,其吸收幅度在红外区的提升最为明显,这表明在X-GaSb体系中,Ni的引入可以有效提升GaSb半导体材料对红外光区及远红外光区光子的响应.图2(b)给出了包含一个X@Sb缺陷体系的光学吸收谱,从图2(b)可以看出,X@Sb缺陷的引入同样可以提升GaSb体系在红外区的吸收幅度,同样也发生了红移现象.比较图2(a)和图2(b)可以看出,X@Sb体系相比于X@Ga体系对红外区甚至远红外光区的光子吸收峰值提升更为明显.总体上来说,过渡族金属的掺杂无论是形成X@Ga缺陷还是X@Sb缺陷都可以改善GaSb体系对红外光区光子的吸收幅度并发生明显的红移现象.
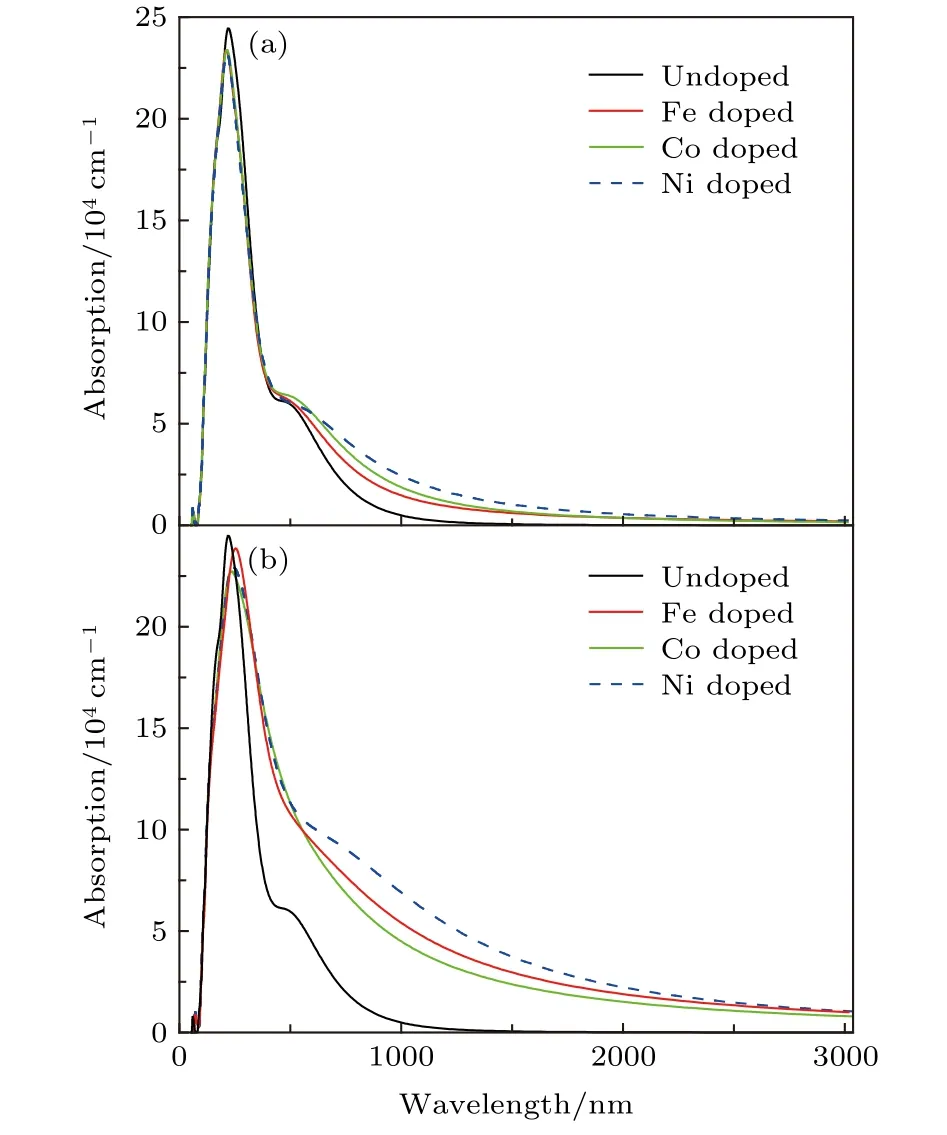
图2 包含(a) X@Ga和(b) X@Sb体系的光学吸收谱Fig.2.Absorption spectra of (a) X@Ga and (b) X@Sb,respectively.
表1列出了Fe@Ga,Co@Ga,Ni@Ga,Fe@Sb,Co@Sb和Ni@Sb这六种缺陷的形成能,形成能的计算公式如下:
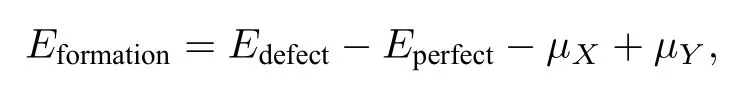
式中Edefect表示包含一个X@Y缺陷的超晶胞的总能量,Eperfect表示完好的GaSb超晶胞的总能量,µX表示过渡金属X的化学势,µY表示Ga原子或Sb原子的化学势.从表1可以看出,X@Sb缺陷的形成能普遍大于X@Ga缺陷的形成能,因此在X掺杂的GaSb体系中,主要以X@Ga缺陷为主.实验上和理论上已经证实[37−40],X@Ga缺陷为主的掺杂GaSb体系是可以稳定存在的,因此本文理论计算模型是合理的.

表1 X@Ga和X@Sb缺陷的形成能Table 1.Formation energy of X@Ga and X@Sb defects.
3.2 X@Ga体系的能带结构和布居分析
以上计算数据分析表明,无论是X@Ga缺陷还是X@Sb缺陷都可以改善GaSb体系对红外光区的吸收幅度并发生明显的红移现象,可以有效提升掺杂体系对红外区光子的响应,但形成能的计算结果表明,X-GaSb体系中主要以X@Ga缺陷为主.因此,本节内容主要讨论X@Ga缺陷对掺杂体系结构的影响以及X@Ga缺陷能带结构.
图3给出了X@Ga体系和未掺杂GaSb体系的能带结构图,红色虚线表示0点费米能级.从图3(a)—(c)可以看出,X的引入将会使GaSb体系的导带底上移,价带顶下移,带隙宽度变大.零点费米能级跨过X引入的杂质能级,使得X-GaSb体系具有了金属相的性质,因此可称为X-GaSb合金材料.图3(a)—(c)的能带结构显示出导带底的下方形成了受主t能级,同时在价带顶上方形成了施主e能级,杂质能级的出现使得掺杂体系的有效禁带宽度变窄并可以作为电子从价带跃迁到导带的桥梁,从而影响了X-GaSb合金体系的光学性质: 电子仅吸收红外光区光子便可从价带跃迁到杂质能级,进一步跃迁至导带,从而提升材料对长波光子的响应,使材料的光学吸收谱的吸收边发生红移现象,提高材料的光催化活性.图3(d)是未掺杂GaSb体系能带结构图,可以看出本征GaSb体系是直接带隙半导体,其带隙宽度理论计算值为0.812 eV,计算结果和实验值符合很好[36].
过渡金属X替代Ga原子形成X@Ga缺陷,掺杂的X原子提供2个4s电子和1个3d电子与周围的4个Sb原子的2p电子成键,因此X原子的五重简并3d轨道上的电子数目变少.X原子周围的4个Sb原子构成四面体,在四面体晶场的影响下,3d轨道电子的简并度将会降低,即五重简并的3d电子轨道将分裂成一个双重简并的e轨道和一个三重简并的t轨道[41].Fe-GaSb合金体系中,Fe@Ga缺陷的电子结构可以表示为e4t1,t轨道能级为未满的,0点费米能级应跨过杂质t能级,可实际上0点费米能级跨过双重简并的e能级,见图3(a),表明Fe原子失电子的数目大于4个.Co@Ga缺陷的电子结构为e4t2,从图3(b)可以看出0点费米能级同样跨过双重简并的e能级,表明Co原子失电子的数目大于5个.Ni@Ga缺陷的电子结构为e4t3,图3(c)中显示0点费米能级跨过三重简并的t能级.以上分析表明,在过渡金属掺杂的GaSb体系,金属X与周围的Sb原子形成的X—Sb键并不是单纯的共价键或者离子键,而是以混合键的形式存在.

图3 X@Ga和未掺杂GaSb体系的能带结构Fig.3.Band structure of X@Ga and undoped GaSb systems,respectively.
为了分析过渡金属X的掺杂对GaSb的影响,给出了掺杂体系和未掺杂体系的Mulliken布居分析,如表2所列.Mulliken电荷(charge)的转移布居可以分析原子的得失电子情况,从表2可以看出,Fe,Co,Ni原子的电荷布居分别为0.45,0.56和0.58,表明三种原子的失电子数目是逐渐增加的,这可以解释图3(a)和图3(b)中0点费米能级跨过双重简并的e能级和图3(c)中0点费米能级跨过三种简并的t能级这一事实.Mulliken键布居(population)可以了解晶体中原子之间的成键特性,从表2可以看出,Fe—Sb,Co—Sb,Ni—Sb三种化学键的键布居分别为0.83,0.80和0.78,表明三种化学键的离子键成分逐渐增强,共价键特性逐渐减弱[42],同时可以看出掺杂体系的键布居数均大于未掺杂体系的布居数,说明掺杂体系的X—Sb键相比于未掺杂体系的Ga—Sb键的共价特性增强,因此X—Sb键的键长都小于Ga—Sb的键长.Fe,Co,Ni三种金属的离子半径小于Ga金属的离子半径,Fe,Co,Ni的掺入将会引起体系的晶格畸变,这种晶格畸变导致X@Ga缺陷内部的正负电荷中心发生分离产生内部的电偶极矩进而产生局域电势差.因此这些过渡族金属的掺杂有利于光生电子-空穴对的分离,可有效提高材料的光催化活性.

表2 GaSb和X掺杂体系的Mulliken布居分析Table 2.Mulliken population of GaSb and X-doped systems.
3.3 X@Ga体系的光学性质
半导体在线性响应范围的光学性质通常用复介电函数ε(ω)=ε1(ω)+ε2(ω) 来描述,其中ε2(ω)表示复介电函数的虚部,由价电子在占据轨道和非占据轨道之间的跃迁来计算,ε1(ω) 表示复介电函数的实部,由ε2(ω) 所满足的Kramers-Kronig色散关系得到[43],方程如下:

其中p为积分主值,

图4为未掺杂GaSb体系和X-GaSb合金体系的复介电函数的实部图,反映了实部随入射光子能量的变化,表征了半导体材料在外电场作用下的极化程度,实部越大表明体系对电荷的束缚能力越强,体系的极化能力越强.在光子能量为0 (无入射光)时,对应的为静态介电常数,从图4可以看出,未掺杂GaSb实部(黑色实线)所对应的数值为22.38,Fe掺杂体系实部(红色实线)所对应的数值为23.95,Co掺杂体系实部(绿色实线)为22.86,Ni掺杂体系实部(蓝色虚线)所对应数值高达48.98.与未掺杂的GaSb体系相比,X-GaSb合金体系的静介电常数都有所增加,说明X掺杂的GaSb体系的极化能力增强,体系的光生电场强度变大,这有利于光生电子-空穴对的迁移和分离,可以有效改善体系的光催化特性,其中Ni-GaSb合金体系的静介电性能最好.

图4 介电函数实部Fig.4.Real part of the dielectric function.
图5为未掺杂GaSb体系和X-GaSb合金体系的复介电函数的虚部图,反映了虚部随入射光子能量的变化.在外加电场下,半导体材料内部形成了大量的电偶极子,虚部表征的是半导体内部形成电偶极子时所消耗的能量,这与电子在能带之间的跃迁有关,反映了半导体材料电子的受激跃迁程度,虚部数值越大表明处于激发态的电子数目越多,进行下一步跃迁的概率也就越大.从图5可以看出,Ni-GaSb体系的虚部(蓝色虚线)在光子低能区(红外光区)远大于Fe,Co掺杂的GaSb体系和未掺杂的GaSb体系,表明Ni-GaSb体系对红外光区光子的吸收能力最大,导致Ni-GaSb体系光学吸收谱的吸收边落在远红外区,其原因在于Ni掺杂在体系0点费米能级附近形成的杂质能级上的电子数目多于Fe,Co掺杂的体系,能够发生能级跃迁的电子数目较多.同时从图5可以看出,X-GaSb合金体系的虚部在光子能量为0处就有很大的响应,其原因在于X引入的e能级或t能级电子是部分占据的,费米能级跨过杂质能级,电子在多重简并的杂质能级之间跃迁几乎不需要能量即可发生,如图3所示.同时从图3可以看出,XGaSb的能带结构图中零点费米能级跨过杂质能级使得GaSb半导体具有了金属相的性质,此时若考虑电子的带内响应对体系介电函数的影响可用基于阻尼振动的Lorentz-Drude模型[44,45]来简单说明.根据电子能带结构的Lorentz-Drude模型在只考虑带内自由电子响应对介电函数的影响时,体系的复介电函数的表达式为

图5 介电函数虚部Fig.5.Virtual part of the dielectric function.

式中ω为入射光频率,Γ为阻尼系数,ωp是金属等离子体频率,ωp>Γ[46].因此当入射光子频率ω→0时εLD(ω) 的实部在远红外光区的数值为负,这不利于掺杂体系内光生“电子-空穴”对的分离.由εLD(ω) 虚部的表达式可知,当入射光子频率ω→0时,其虚部的数值趋向于无穷大,这表明入射光波波长较长时不会发生自由电子的能带之间的直接跃迁效应,电子的吸收效应起主要作用.实际上自由载流子吸收过程联系的是同一个带内电子状态之间的跃迁,载流子的吸收是一个间接跃迁的二级过程,在自由载流子吸收光子的同时伴随有声子的散射和电离杂质的散射,电子的带内响应并不会产生有效的“电子-空穴”对,因此不利于材料光学性质的改善.电子发生间接跃迁过程的概率要比电子与电磁波相互作用的直接跃迁的概率小得多,因此自由电子的带内响应效应对材料光吸收系数的贡献要小得多[47].由X-GaSb引入的杂质能级上自由电子的带内响应对GaSb半导体的光学性质有一定的影响,但不会太大,起主要作用的仍然是电子带间的直接跃迁.
图6给出了未掺杂GaSb体系和X-GaSb合金体系的反射光谱.利用复折射率与复介电函数之间的关系((n(ω)+ik(ω))2=ε1(ω)+iε2(ω))和Kramers-Kronig关系可以得到吸收系数α(ω) 和反射系数R(ω) 的表达式为[43]

式中c为光速;n=n(ω) 为复折射率的实部,就是普通折射率;k=k(ω) 是复折射率的虚部,决定了光的衰减,与吸收系数α(ω) 直接有关.由上式可以看出,吸收系数α(ω) 和反射系数R(ω) 都是k的单调增函数,因此吸收系数大的介质其反射系数也大,如果一种固体强烈吸收某一光谱范围内的光,它就能有效地反射在同一光谱范围内的光.XGaSb合金体系具有金属化性质,当入射长波光子时,带内的自由电子气极易被低频交变电场极化从而对光场的电磁屏蔽作用很强,这一特性使得自由电子气模式材料在入射光子能量在红外和远红外区的位置具有很高的反射率.从图6可以看出,XGaSb合金体系在红外光区的反射率高于未掺杂的GaSb体系.综合图2和图5可知,X-GaSb合金体系对红外和远红外区的光子有强烈的吸收,对红外光区的利用率最高,X的掺入可以有效提高X-GaSb合金材料的光催化特性.其中Ni-GaSb合金体系在红外光区的数值(蓝色虚线)远大于Fe,Co掺杂的GaSb体系,表明Ni掺入对改善GaSb材料的光催化特性最优.
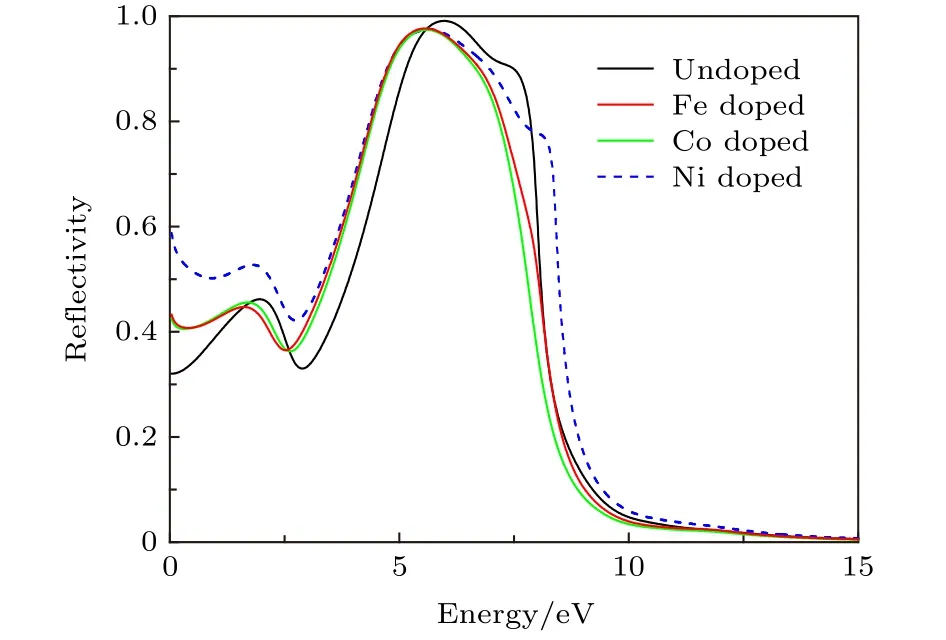
图6 反射光谱Fig.6.Reflectivity of GaSb and X-doped systems.
3.4 不同浓度Ni-GaSb合金体系的光学性质
从以上分析可以看出,X的掺杂对GaSb体系的光学性能都有很大的改善,表现为掺杂体系的极化能力增加,对红外光区光子的响应能力增强,掺杂体系光学吸收谱的吸收边发生明显红移,由掺杂的金属离子引起的晶格畸变而建立的内置电场可以有效提高光生电子-空穴对的迁移和分离,进而提升材料的光催化性能等.考虑到Ni的掺入对体系光学性质的改善最优,本节主要讨论了不同浓度Ni-GaSb合金体系的光学性质,期望找出最优的Ni原子掺杂比例.
图7给出了不同掺杂Ni原子浓度下GaSb体系的光学吸收谱,对于不同浓度的掺杂体系,使Ni原子的分布尽可能均匀.从图7(a)和图7(b)可以看出,随着掺杂Ni原子数目的增加,体系在红外光区的吸收幅度逐步增加,当掺杂Ni原子的数目为7个时达到最大.图7(c)则表明,进一步增加掺杂Ni原子的数目,体系的吸收幅度又逐步降低,因此我们认为在包含64个原子的GaSb超晶胞合金中,替代Ga原子的Ni原子为7个时,体系对红外光区光子的响应最好,此时Ni原子的掺杂摩尔浓度为10.94%.

图7 不同浓度Ni掺杂的光学吸收谱Fig.7.Optical absorption spectra of different Ni-doped density.
7个Ni原子的掺杂属于高浓度掺杂,掺杂Ni原子可替代GaSb超晶胞合金中Ga原子的多个位置,因此有必要考虑不同掺杂位置对体系光学性质的影响.为了描述的方便,用阿拉伯数字来标记超晶胞中被替代Ga原子的不同位置(见图1).考虑到体系的对称性和计算体系的周期性边界条件,设计了四种不同的掺杂结构: 当用7个Ni原子分别替代图1中1,2,3,4,5,6,12号Ga原子时构成结构S1=S(1,2,3,4,5,6,12),同样的方式分别得到结构S2=S(1,3,4,5,6,8,9),S3=S(1,3,5,6,7,8,12)和S4=S(3,6,7,10,11,12,13).从图1可知,这4种结构中Ni原子的密集程度逐渐增加,其中结构S1为7个Ni原子均匀掺杂的情形.表3列出了这四种掺杂结构优化后的体系的总能量.从表3可知,对于均匀掺杂的结构S1体系的总能量最小,在这种结构中,掺杂的Ni原子之间的距离比较远,Ni-Ni之间的相互作用较弱.

表3 S1,S2,S3和S4掺杂结构优化后体系的总能量Table 3.Total energies of relaxed S1,S2,S3 and S4 configurations.
图8给出了这4种结构体系的光学吸收谱.从图8可以看出,均匀掺杂情形下的S1结构体系(蓝色虚线)在红外光区的吸收幅度最大,随着掺杂Ni原子密集程度的增加,光学吸收幅度逐步降低.密集掺杂的S3和S4结构体系在红外区的曲线几乎重合,光学吸收谱的吸收幅度最低.由Ni原子引入深杂质能级的附加势能是作用距离仅为一两个原子间距的短程势[43],因此密集掺杂的S3和S4结构中的Ni原子可以成为“电子-空穴”对的有效复合中心,从而使载流子的寿命大大降低,影响材料的光学性能.由此可知,半导体材料的光学特性不仅和掺杂元素的种类和浓度有关,也和掺杂元素的均匀程度有关.表3的数据表明均匀掺杂的S1结构体系的总能量最低,说明在高浓度掺杂GaSb合金体系中Ni原子不易形成团簇结构,另外在分子束外延生长过程中,通过控制生成氛围可以实现动力学控制下的异质外延生长,对于高浓度均匀掺杂的S1结构,在实验上的实现并不困难[39,40].因此S1结构是改善GaSb材料光学性能的最优掺杂结构.

图8 S1,S2,S3和S4结构体系的光学吸收谱Fig.8.Optical absorption spectrum of S1,S2,S3 and S4 configuration.
4 结 论
运用第一性原理LDA+U的方法研究了XGaSb合金体系的电子结构和光学性质,结论如下:1)X的掺入主要形成X@Ga和X@Sb两种缺陷结构,形成能的计算结果表明X-GaSb合金体系主要以X@Ga缺陷形成存在,且两种缺陷结构的存在都可以有效提升GaSb体系在红外光区的光学吸收幅度; 2)X@Ga缺陷体系的能带结构表明,X的掺杂可以在0点费米能级附近引入杂质能级并使掺杂体系的有效禁带宽度变窄.布居分析表明Fe,Co,Ni原子的失电子数目逐步增强,X—Ga键的布居数逐步变小,说明杂质的掺入对GaSb体系的晶格畸变逐步增强,有利于光生电子-空穴对的分离,提高GaSb材料的光催化特性; 3) 与未掺杂的GaSb相比,所有掺杂体系的静介电常数均有很大增加,说明掺杂GaSb体系的极化能力增强,掺杂体系的虚部数值在红外光区的数值变大,表明X的掺杂可以有效提高GaSb材料对红外区光子的吸收,0点费米能级跨过杂质能级是掺杂体系复介电函数虚部在光子能量为0时就有响应的原因.掺杂体系的反射光谱同样表明了X的掺入对GaSb体系的光学性能有很大的影响; 4) 综合分析表明,X的掺杂对GaSb体系的光学性能都有很大的改善,但Ni掺入对改善GaSb材料的光催化特性最优;5) Ni-GaSb合金体系的光学性能不仅与掺杂浓度有关,还与掺杂的均匀程度有关.均匀掺杂可以避免光生电子-空穴复合中心的形成,通过计算得出最佳的Ni原子掺杂摩尔浓度为10.94%,此时光学吸收范围和吸收峰值都达到最大,有效提高了材料的光催化性能.期望我们的研究可扩展GaSb材料在红外热光伏电池,红外光探测器和红外半导体激光器等领域的应用.
