TANK结合酶1小分子抑制剂的研究进展
2019-09-23常玉丁克
常玉,丁克
(暨南大学药学院,广东 广州 510632)
TANK结合激酶1(TANK binding kinase 1,TBK1)是一种丝氨酸/苏氨酸蛋白,属于非经典的IκB激酶(IKK)家族。TBK1通过调控干扰素调节因子(IRF)、核因子κB(NF-κB)等多条信号通路和转录因子,在免疫、肿瘤、炎症、代谢等疾病的发生和发展过程中发挥重要作用。作为肿瘤免疫治疗的重要潜在靶标之一,近年来对TBK1的作用机制及小分子抑制剂的研究逐渐成为热点。本文针对TBK1在肿瘤免疫中的生物机制以及小分子抑制剂研究进展进行综述。
1 TBK1的蛋白结构和信号通路
IKK蛋白家族可分为经典和非经典2类亚型蛋白。经典的IKK蛋白包括IKKα、IKKβ、IKKγ(又称NEMO);非经典的IKK蛋白包括IKKε和TBK1。TBK1与IKKε的氨基酸序列具有较高相似性,其序列比对同一性高达49%,相似性65%,二者具有多种相似的生物学功能[1-2]。尽管如此,二者的体内分布却明显不同,TBK1与经典的IKKα、IKKβ一样,在体内各个组织和器官中广泛表达,而IKKε主要表达于淋巴组织、外周血淋巴细胞和胰腺[3]。另外,小鼠和人类的TBK1蛋白具有超过99%的同源性,说明该蛋白在哺乳动物中高度保守[2]。
TBK1蛋白由729个氨基酸组成,包含4个主要的结构域:位于N-端的激酶域(kinase domain,KD,9 ~ 301)、位于C-端的亮氨酸拉链域(leucine zipper,LZ,499 ~ 527)、螺旋-环-螺旋区(helixloop-helix motif,HLH,591 ~ 632)和泛素样结构域(ubiquitin-like domain,ULD,305~383)(见图1)[1]。LZ和HLH对TBK1蛋白的二聚化激活至关重要[4];ULD是TBK1蛋白的主要调节区域,调控蛋白的激活、底物结合以及下游信号通路[5]。TBK1通过激酶域第172位丝氨酸磷酸化而被激活[6],激活后的TBK1蛋白中Ser172附近loop区发生较大变化,导致αC螺旋向口袋内发生较大扭转(见图1),后口袋闭合,而蛋白其他部分构象不发生明显改变[6-7]。

图1 TBK1蛋白结构Figure 1 Crystal structure of TBK1
研究人员最早在小鼠体内发现,TBK1蛋白通过与TANK、TNF受体相关因子2(TRAF2)形成三元复合物来激活NF-κB信号通路,同时,该三元复合物的形成对TBK1蛋白活化也至关重要[2]。另外,在对下游干扰素诱导的IRF家族转录因子的激活过程中,非经典的IKK蛋白TBK1和IKKε,比经典的IKK蛋白(IKKα和IKKβ)具有更为重要的调节作用。TBK1通过与其类似蛋白IKKε形成同源二聚体进而被激活,TBK1/IKKε复合物是下游干扰素β(IFN-β)、前列腺素E2(PGE2)、一氧化氮(NO)等转录因子表达的重要调控因子[8-9]。TBK1在多条细胞信号通路,包括转录因子NF-κB、IRF3通路,以及Ⅰ型干扰素(IFN-I)、Ⅱ型干扰素(IFN-II)靶基因的调控中发挥关键作用,与多种疾病,特别是免疫、自噬、代谢、炎症以及肿瘤的发生发展密切相关[10]。
当病原体侵入后,模式识别受体(PRRs),如Toll样受体4(TLR4)、视黄酸诱导基因样受体(RLRs)和胞浆DNA受体等先天免疫传感器与其同源配体,如脂多糖(LPS)、双链RNA或DNA等相互作用,招募TRIF(TIR domain-containing adaptor-inducing IFN-β) 和 TRAF3, 促使 TBK1 与其类似蛋白IKKε或NF-κB活化激酶相关蛋白1(NAP1)形成复合物,最终被激活[11]。活化的TBK1进而使IRF3磷酸化,导致其同源二聚化并转移至细胞核,在细胞核内诱导抗病毒IFN-I和IFN-II表达[12-14],从而警告邻近细胞(包括免疫细胞)危险和外来入侵,形成早期的防御反应(见图2)。同时,活化的TLR3也能够招募其配体TRIF、TRAF3激活TBK1,从而诱导干扰素产生。除细胞膜上的TLRs蛋白外,由病毒RNA活化的胞浆RLRs、双链DNA活化的胞浆DNA受体环状GMP-AMP合成酶(cGAS)以及干扰素基因刺激蛋白(STING)启动子信号[15-18],都能够激活下游TBK1蛋白继而诱导IRF3活化,在某些情况下也能够诱导IRF7的活化,诱导干扰素产生[19-20](见图2)。另外,TBK1能够与同源蛋白IKKβ二聚化,激活下游NF-κB转录因子p50、p65,从而导致TNF-α、IP-10等多种炎症因子产生。此外,天冬氨酸-谷氨酸-丙氨酸-天冬氨酸(DEAD)解螺旋酶3(DDX3X)也被证实在DNA和病毒RNA识别后能够直接与小鼠巨噬细胞中的TBK1相互作用,诱导 IFN-β 表达[21]。
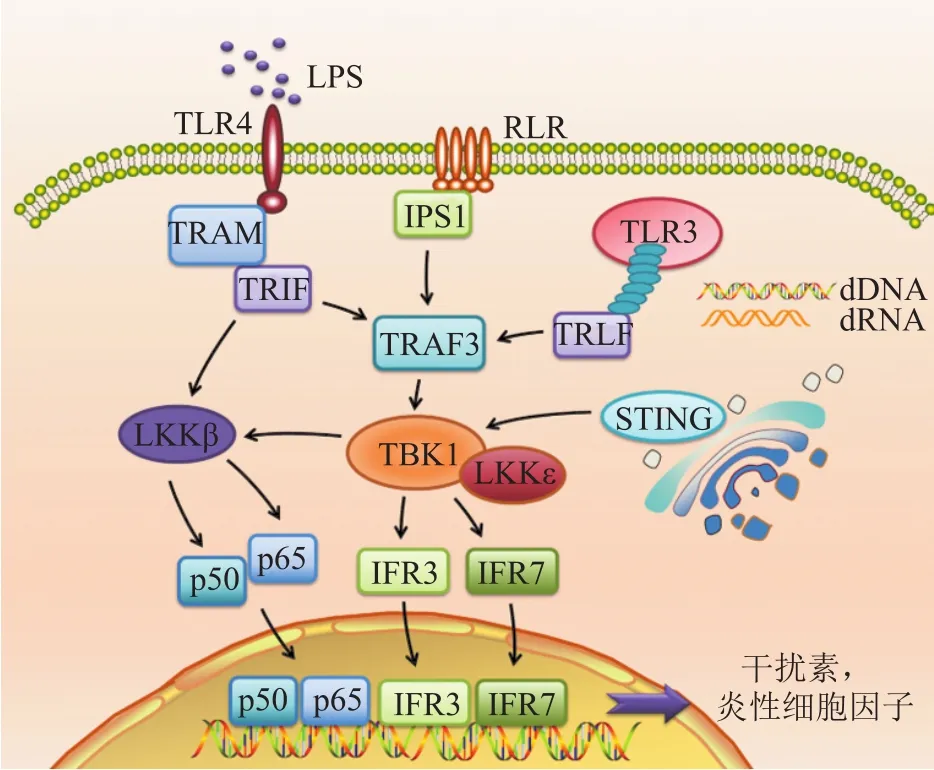
图2 TBK1介导的信号通路Figure 2 TBK1-mediated signaling pathways
2 TBK1与肿瘤发生发展
研究表明,TBK1蛋白在多种肿瘤的发生发展中发挥重要作用[22-24]。Barbie等[25]发现TBK1和其诱导的NF-κB信号传导在KRAS突变肿瘤生长中是必需的。在人肺癌细胞中,对TBK1诱导凋亡的抑制作用依赖于Kras致癌基因的表达,TBK1是KRAS致癌的重要合成致死因子。此外,TBK1在黑色素瘤和非小细胞肺癌(NSCLC)中也具有致癌作用[26-27]。Eskiocak等[26]在对耐药突变的黑色素瘤研究中发现,含有BRAF突变的对威罗非尼和曲美替尼耐药的黑色素瘤细胞对TBK1/IKKε蛋白较敏感,TBK1/IKBKε抑制剂能够选择性地抑制含有BRAF耐药突变的黑色素瘤细胞活性。在黑色素瘤中,NRAS过表达会促进TBK1磷酸化激活。利用siRNA将5种不同的NRAS突变黑色素瘤细胞中TBK1敲除,能够有效地抑制黑色素瘤细胞的迁移和侵袭,相反地,持续高表达TBK1导致侵袭增加,然而在NRAS野生型黑色素瘤细胞中没有这种现象。此外,NSCLC的部分癌细胞也表现出对TBK1抑制的敏感性,这与Akt和mTORC1信号通路的激活相关[27]。在人T细胞白血病1型病毒(HTLV-1)诱导的成人T细胞白血病(ATL)的研究中发现,TBK1/IKKε对STAT3蛋白在HTLV-1转化的T细胞中的激活至关重要,将TBK1或IKKε沉默均可导致STAT3失活,抑制白血病细胞生长[28]。因此,TBK1/IKKε在HTLV-1转化的T细胞的生长、增殖中具有关键作用。在乳腺癌中,TBK1能够通过对ERα第305位丝氨酸进行磷酸化修饰,增加ERα的转录活性,最终导致雌激素耐药产生[29]。
功能复杂的蛋白激酶TBK1能够通过介导多条途径来驱动肿瘤生长,主要包括:TBK1与有丝分裂底物PLK1、CEP170和NUMA相互作用,促进癌细胞的细胞分裂,进而促进微管稳定性和有丝分裂;TBK1诱导癌细胞自噬,抑制引发免疫反应的多种促炎信号;TBK1还能够参与Akt/mTOR信号通路的激活,促进癌细胞存活。
2.1 TBK1与有丝分裂
据报道,TBK1能够通过直接磷酸化Akt来激活polo样激酶1(PLK1)[27,30]。然而在人肺癌A549细胞中,敲除TBK1蛋白能抑制A549细胞生长,但不影响Akt蛋白活性[31]。进一步对磷酸化蛋白的质谱分析表明,TBK1的敲除的确能够抑制PLK1磷酸化激活。研究还发现TBK1在体外能够诱导PLK1磷酸化,TBK1磷酸化在有丝分裂期间增加并位于中心体、有丝分裂纺锤体和中间体,对TBK1的选择性抑制或敲除能够阻断PLK1有丝分裂相关蛋白的磷酸化激活,引起纺锤体组装缺陷,抑制有丝分裂的进行[31]。TBK1能够与中心体蛋白CEP170和有丝分裂器蛋白NuMA结合,二者均为TBK1的底物。在CEP170中心体定位、CEP170与微管解聚酶Kif2b结合以及NuMA与动力蛋白结合过程中,TBK1发挥着关键作用[32]。此外,选择性破坏TBK1-CEP170复合物能够增加微管稳定性,引起有丝分裂缺陷,这表明TBK1是微管动力学和有丝分裂所必需的有丝分裂激酶。
2.2 TBK1与自噬
自噬是由饥饿反应诱导的在溶酶体中捕获并降解细胞内蛋白质和细胞器,回收细胞内组分以维持细胞代谢和存活的过程。自噬在控制细胞内蛋白质、细胞器的质量和数量方面起着重要的稳态作用,当自噬功能失调时,会诱发多种疾病,包括癌症[33-34]。自噬在癌症中的作用机制很复杂,在不同的情况下分别发挥中性、促进或抑制肿瘤生长的不同生理功能,具体取决于微环境中的营养物、微环境胁迫以及免疫系统等[29]。研究表明:自噬能够通过抑制p53通路,防止能量危机、细胞死亡和衰老,抗肿瘤免疫等途径促进癌症的发生发展;相反地,在肿瘤发生初期,自噬能够抑制肿瘤形成,例如在胰腺癌中,自噬能够阻止炎症反应和细胞损伤,从而抑制胰腺癌的发生发展[35]。
TBK1能够通过多种方式参与自噬。研究表明:TBK1能够诱导自噬受体视神经蛋白第177位丝氨酸磷酸化激活,促进细胞内细菌的清除[36-37]。另外,TBK1通过对p62/SQSTM1第403位丝氨酸磷酸化来调控自噬以及线粒体的自噬体吞噬[38-39],而p62/SQSTM1被证实与癌症的发展相关[40]。Wild等[36]发现被TBK1激活的p62/SQSTM1能够通过降解STING,抑制天然免疫反应。有研究表明:在巨噬细胞中,TBK1调控p62磷酸化水平,p62是自噬清除中所必需的调控因子,在自噬成熟过程中发挥关键作用[41]。同时,促炎细胞因子白细胞介素1β(IL-1β)能够诱导巨噬细胞中分枝杆菌的自噬清除,而该细胞因子IL-1β活性也依赖于TBK1。Yang等[42]发现,在自噬缺失的胰腺炎小鼠模型中,TBK1蛋白磷酸化水平增加,中性粒细胞、T细胞渗透性以及PK-L1蛋白也随之上调和增加。在体外胰腺导管腺癌细胞中,由自噬抑制引起的TBK1蛋白的过度激活也能够诱导CCL5、IL-6蛋白表达上调(促进肿瘤发生)以及多种T细胞和中性粒细胞趋化因子的增加。相应地,自噬对pTBK1的降解也限制了TBK1信号传导,防止TBK1过度激活自噬并限制促炎细胞因子的产生以及中性粒细胞、T细胞的募集。自噬与TBK1之间存在负反馈调节,活化的TBK1促进PDA细胞中的基础自噬,然后在自噬过程中被降解,以限制自噬和TBK1诱导的细胞因子产生的过度激活,二者都促进肿瘤形成。研究还发现,TBK1/IKKε/JAK抑制剂CYT387(momelotinib)不仅抑制自噬,还能够抑制这种反馈炎症并降低PD-L1表达,限制KRAS驱动的胰腺异常生长[42]。
2.3 TBK1与肿瘤免疫
天然免疫感应是促进肿瘤中T细胞产生和渗透性的关键步骤。TBK1被广泛认为是天然免疫激酶,作为IFN-I反应通路中STING的跨膜蛋白刺激物的下游蛋白。细胞内病原体感染的核酸引起STING活化,进而逐级启动下游信号,包括TBK1介导的IRF3和Stat6等,最终导致IFN-I和细胞因子产生[19,43]。在小鼠模型中,STING的激活能够通过诱导肿瘤微环境中IFN-β的产生,发挥抗肿瘤免疫作用。在B16黑色素瘤、4T1乳腺癌以及CT26结肠癌的小鼠模型中,瘤内注射STING激动剂环状二核苷酸衍生物均能够诱导IFN-β、细胞因子和CD8+T细胞产生,抑制原发性肿瘤生长并使其消退,同时也抑制远端病变的发生,促进免疫记忆的系统性免疫应答[44]。Woo等[44]在该工作中强调了天然免疫调节策略的抗肿瘤功效,并暗示TBK1能够作为潜在的抗肿瘤蛋白。
与上述研究结果相反地,Xiao等[45]发现在树突状细胞中,TBK1能够促进肿瘤生长,是抗肿瘤免疫的潜在靶标。他们通过构建选择性敲除树突状细胞中TBK1的小鼠模型(TBK1-DKO)与野生型小鼠(WT)对比发现,TBK1为树突状细胞的非必需蛋白,TBK1的特异性缺失会破坏T细胞稳态,使T细胞活化并引发自发性自身免疫;同时,皮下注射肿瘤细胞的TBK1-DKO小鼠的寿命更长,肿瘤更小,提示TBK1的特异性缺失还增强了小鼠模型中的抗肿瘤免疫力。对TBK1-DKO和WT小鼠的骨髓和脾脏对比表明,二者具有相似的外周免疫谱,说明TBK1对骨髓细胞发育不是至关重要的。然而,含有B16黑色素瘤的抗肿瘤免疫研究中,与WT小鼠相比,TBK1-DKO小鼠对肿瘤和淋巴结表现出更强的T细胞效应以及抗PD-1治疗的协同作用,并且在另外2种肿瘤细胞(EG7-OVA和EL4淋巴瘤细胞)也证实了这些结果。进一步研究表明,TBK1能够反向调节Ⅰ型干扰素受体(IFnAr)诱导的下游基因,敲除IFnAr1能够抑制TBK1-DKO小鼠中异常的T细胞活化和自身免疫反应。这些研究结果证实了TBK1在树突状细胞中的促肿瘤功能,TBK1通过抑制IFnAr1信号传导以介导免疫耐受,促进肿瘤生长。
尽管上述2篇文献对TBK1在肿瘤免疫中的作用解释相互矛盾,但可以发现TBK1的生物功能在不同的癌症类型以及含有肿瘤的不同基质细胞类型中都可能是不同的,目前对TBK1在肿瘤免疫研究中的具体作用机制尚未完全明确,针对评价TBK1在某一种细胞类型和特定肿瘤中对肿瘤细胞生长的作用机制研究可能为该领域的研究方向之一。
3 TBK1小分子抑制剂
TBK1在肿瘤、免疫、自噬等疾病中发挥重要作用,是多种疾病治疗的重要潜在靶标,因此,寻找TBK1小分子抑制剂具有重大意义:一方面可以为探究TBK1的复杂生物学功能提供小分子探针工具;另一方面也为相关疾病的治疗提供药物候选化合物。近年来对TBK1小分子抑制剂的报道逐渐增多,但仍非常有限。已报道的TBK1小分子抑制剂大多具有类似骨架,即2-氨基嘧啶类衍生物。尽管一些化合物对TBK1表现出较好的体外、体内活性以及选择性,但进一步寻找高效的选择性TBK1小分子抑制剂仍十分必要。
3.1 2-氨基嘧啶类TBK1小分子抑制剂
BX795(1)是最早被发现并申请专利保护的代表性TBK1小分子抑制剂[46],最初作为PDK1抑制剂设计而被报道[47],在进一步激酶活性筛选时发现,BX795对多种蛋白激酶均表现出高抑制活性,其中对 TBK1 的激酶活性(IC50= 2.3 nmol · L-1)比对原靶标PDK1的活性(IC50=7 nmol · L-1)还要强;此外,对 TBK1 同源蛋白 IKKε(IC50= 9.5 nmol · L-1)和Aurora B(IC50=11 nmol · L-1)等表现出高抑制活性,类似化合物BX320(2)对TBK1的抑制活性与BX795相当[48]。进一步在口腔鳞状细胞癌(OSCC)细胞中的作用机制研究发现,BX795能够抑制Akt和NF-κB信号转导,阻止细胞进入有丝分裂期,并增加自噬癌细胞的产生,从而剂量依赖性地抑制癌细胞增殖[49]。Tu等[50]成功解析了BX795与TBK1蛋白的晶体结合模式(见图3),BX795结合于TBK1蛋白的ATP结合口袋并形成多组相互作用。在铰链区,BX795的1-N和4-NH能够与Cys89形成2个氢键,8'-NH与β1折叠上Leu15的C = O形成氢键,BX795中9'位C = O分别与Ser93和Thr96形成3组氢键作用,在后口袋部分,BX795第6''位C = O能够与Lys38形成一对氢键。BX795既可以结合于未活化的TBK1蛋白,也能够与活化的TBK1蛋白结合。随后以BX795结构为基础,研究人员又陆续优化、报道了多种2-氨基嘧啶类TBK1小分子抑制剂。
英国邓迪大学的Clark等[51]对BX795进行结构优化得到化合物MRT67307(3),其具有良好的激酶抑制活性,对TBK1和IKKε的IC50分别为19和160 nmol · L-1,而对同源蛋白IKKα和IKKβ未表现出明显的抑制活性;与BX795不同,MRT67307对JNK和p38 MAPK等蛋白也不具有抑制活性,展现了较好的选择性。在解析的MRT67307与TBK1蛋白晶体结构中,2-氨基嘧啶的结构保持了化合物在铰链区与Cys89形成的2个氢键作用,第6''位C = O与Thr156形成氢键,尾部的环丁基被DFG区域包裹(见图4)[50]。与BX795结构头部不同,MRT67307头部甲基吗啉的结构伸向溶剂域,未与蛋白形成多个氢键作用,这可能是导致MRT67307较BX795活性低的原因。

图3 BX795与TBK1共晶结构(PDB: 4jl9)Figure 3 Co-crystal structure of BX795 with TBK1(PD: 4jl9)
Richters等[52]利用活性筛选的方法获得了多个含有嘧啶结构的TBK1抑制剂,经过一系列结构优化发现,在嘧啶结构第2、4、6位带有取代基的骨架结构有助于提高TBK1抑制活性,并优选出化合物 4 ~ 7,其对 TBK1 的 IC50为 0.06 ~ 0.38 μmol · L-1,其中以含有强吸电子基团硝基的化合物4活性最优;同时这类化合物也展现出较好的体外抗炎活性。
McIver等[53]报道了一系列BX795衍生物(8 ~15)作为TBK1选择性抑制剂。通过改变5位取代基团,如溴、碘、脲、芳基酰胺、N-甲基哌啶等,开发了40多种不同的5-取代-2,4-二氯嘧啶中间体。其中,化合物11对TBK1抑制活性最强,IC50达2 nmol · L-1。化合物15也具有高抑制活性(IC50=8 nmol · L-1)和选择性,并且在人和小鼠肝微粒体中表现出较好的稳定性。此外,化合物15还能够抑制小鼠体内LPS诱导的促炎细胞因子释放。
Scripps研究所报道了以2-氨基-4-(3-氰基-40-吡咯烷)苯基-嘧啶的刚性结构为骨架的一类2-氨基嘧啶类TBK1小分子抑制剂[54]。他们以JNK抑制剂SR8185(16)为先导化合物,通过系统的构效关系研究和结构优化成功获得对TBK1具有高抑制活性和高选择性的化合物17和18,其IC50均小于1 nmol·L-1。化合物17和18在人乳腺癌、前列腺癌和口腔癌细胞中都表现出良好的细胞抑制活性,并且在异种移植和同种异体移植小鼠模型中能够显著抑制肿瘤的生长,显示出较好的体内抗肿瘤活性。此外,化合物17和18具有低相对分子质量、低细胞色素P450抑制和高代谢稳定性等成药特性,有望成为TBK1抑制剂进入临床试验的潜在候选化合物。

图4 MRT67307与TBK1共晶结构(PDB: 4iwq)Figure 4 Co-crystal structure of MRT67307 with TBK1 (PDB: 4iwq)

Domainex是研发TBK1/IKKε激酶抑制剂较早的药物研发公司之一,他们设计开发了80余个含有2-氨基嘧啶的TBK1/IKKε激酶抑制剂(如19~22),其中多个化合物具有较好的口服生物利用度、强效的体外激酶抑制活性,并且在动物疾病模型中也表现出较好的体内活性,可用于治疗TBK1相关疾病[55]。特别地,化合物20和21对TBK1具有高激酶抑制活性,IC50达到 1~2 nmol·L-1,且对其他相似蛋白,如IKKβ、JNK-1、JNK-3表现出200倍以上的选择性。有趣的是,在进一步进行的炎症小鼠模型试验中,该类化合物能够抑制多种促炎细胞因子,包括TNF-α、RANTES、IL-1β、IL-6,且不会出现副作用。这些结果表明该类化合物有望作为治疗TBK1相关炎症等疾病的药物候选化合物进行进一步的研究开发。

Myrexis公司是研发TBK1抑制剂领先的药物公司之一,设计合成了超过670个2-氨基嘧啶类化合物(如23~28)[56],其中大量化合物具有良好的TBK1抑制活性,IC50小于 10 nmol·L-1。代表性化合物MPI-0485520(23)对TBK1具有强效的抑制活性和选择性,IC50达到500 pmol·L-1。多个化合物在炎症小鼠模型中表现出良好的体内外抑制活性,能够显著抑制RANTES、IP-10和IFNβ蛋白表达。例如,化合物26~28对RANTES的IC50均小于10 nmol·L-1,化合物26对IP-10和IFNβ的IC50分别为60和40 nmol·L-1,表明该类化合物对类风湿性关节炎和其他相关疾病的患者具有潜在的治疗效果。
最近,Thomson等[57]报道了一个新型的2-氨基嘧啶类选择性TBK1小分子抑制剂GSK8612(29),该化合物具有良好的细胞活性,能够抑制淋巴瘤细胞Ramos中TLR3诱导的IRF3磷酸化、人原代单核细胞中IFN-I的分泌,以及人淋巴瘤细胞THP1中IFN-β的产生。同时,GSK8612在多种细胞中未发现显著的脱靶靶点,表现出较好的TBK1选择性,能够作为探针分子进一步探究TBK1在免疫、神经炎症、肥胖及肿瘤中的生物学功能。

3.2 3H-咪唑并[4, 5-b]吡啶类TBK1小分子抑制剂
2012年阿斯利康公司报道了44个3H-咪唑并[4,5-b]吡啶类TBK1小分子抑制剂[58],几乎所有这些化合物在1 μmol · L-1浓度下均显示出对TBK1的激酶抑制活性,多个化合物(30 ~ 37)IC50均小于10 nmol · L-1。代表性化合物33、35、36对TBK1激酶表现出强抑制活性,其IC50分别为4、3 和4 nmol · L-1,但这3个化合物缺乏对其他激酶如IKKε、Aurora B和CDK2的选择性。化合物37对TBK1显示出较好选择性,对 TBK1、IKKε的IC50分别为9 和 46 nmol ·L-1,而对Aurora B和CDK2没有明显的抑制活性。

2014年,阿斯利康公司继续报道了另外35个作为TBK1小分子抑制剂的3H-咪唑并[4, 5-b]吡啶类衍生物(如化合物38 ~ 41)[59]。通过引入噻唑基团所得的化合物41对TBK1具有强抑制活性,IC50为9 nmol · L-1,而对 IKKε、Aurora B 和 CDK2 的 IC50分别为 42 nmol · L-1、1.45 μmol · L-1和 0.284 μmol · L-1,表现出对TBK1较强的抑制活性和选择性。化合物39也具有较好的选择性,对TBK1、IKKε和Aurora B 的 IC50分别为 77 nmol · L-1、0.273 μmol · L-1和 5.92 μmol · L-1,对 CDK2 则没有明显抑制作用。

3.3 Amlexanox类TBK1小分子抑制剂
Amlexanox(42)是美国FDA批准上市的治疗哮喘和口腔溃疡的药物,2013年被确定为TBK1和IKKε抑制剂,其对 2 种激酶的 IC50为 1 ~ 2 μmol · L-1[60-61]。在肥胖小鼠模型中,amlexanox能够显著影响小鼠体内代谢功能,恢复cAMP对儿茶酚胺的敏感性,激活p38丝裂原活化蛋白激酶(MAPK),诱导脂肪组织中解偶联蛋白1(Ucp1)的表达,通过增加能量消耗导致体质量减轻,同时,amlexanox还能够通过IL-6激活肝JAK/STAT通路,改善胰岛素敏感性,调节葡萄糖水平[62]。鉴于amlexanox在临床使用中具有良好的安全性,Oral等[63]在2型糖尿病和非酒精性脂肪肝患者中进行了临床试验,结果证实amlexanox能够改善糖尿病患者的血糖水平,部分患者表现出肝脏脂肪减少的治疗效果,这可能与能量消耗增加有关。最近,Beyett等[64]设计合成了一系列amlexanox衍生物(43 ~ 47)作为TBK1/IKKε选择性抑制剂,多数衍生物对TBK1表现出微摩尔级的激酶抑制活性。他们重点探讨了3位取代基对活性的影响,并成功解析了amlexanox以及化合物43、45、46与TBK1蛋白的晶体复合物结构(见图5)。与2-氨基嘧啶类抑制剂类似,在TBK1蛋白铰链区,amlexanox中1-N、2-NH2也分别与Cys89、Glu87形成2个氢键,3位羧基对活性保持很重要,当成酯或变成酰胺后活性下降,如化合物43对TBK1的活性下降为amlexanox的1/70,其IC50为55 μmol · L-1。当 3 位羧基替换成四氮唑(化合物 46)时,能够与 Met86 形成氢键作用,IC50为 0.4 μmol · L-1,比 amlexanox(IC50=0.8 μmol · L-1)活性提高 1 倍。


图5 化合物42、43、45和46与TBK1共晶结构Figure 5 Co-crystal structures of compounds 42, 43, 45 and 46 with TBK1
3.4 TBK1小分子蛋白降解剂
蛋白降解靶向嵌合体(proteolysis-targeting chimaera,PROTAC)技术是将靶蛋白配体与E3连接酶配体连接形成双功能分子,募集E3连接酶至靶蛋白以促进其泛素化,进而被蛋白酶体系统识别并降解。PROTAC在药物研发中具有其独特优势,近年来已成为热点研究领域。最近,Crews等[65]已成功将PROTAC技术应用于TBK1靶向降解。他们将2-氨基嘧啶类TBK1小分子配体48/49与E3连接酶VHL配体(50)连接起来,并对TBK1配体和连接链进行了系统的构效研究,优化得到的化合物51能够显著地剂量依赖性地降解TBK1 蛋白(DC50=12 nmol·L-1,Dmax=96%),而不影响同源蛋白IKKε表达,具有高选择性。特别地,在多种含有KRAS突变型和野生型细胞,包括H23细胞、A549细胞、H1792细胞、H2110细胞和HCC827细胞中,化合物51均能够显著抑制TBK1蛋白表达。该化合物为进一步研究TBK1相关生物学功能提供了重要的小分子探针工具。

4 总结与展望
TBK1蛋白参与肿瘤发生发展、免疫反应、自噬、炎症、代谢等多种生理学过程。尽管有研究表明TBK1在黑色素瘤、NSCLC、乳腺癌等肿瘤生长中起关键作用,但其过程涉及对细胞周期、自噬、免疫等诸多方面影响,分子作用机制尚未完全明确,还需进一步细化研究TBK1在不同的疾病、不同细胞类型中的潜在生物学功能及作用机制。目前针对TBK1抑制剂研究,除amlexanox获批用于临床治疗哮喘及口腔溃疡外,尚无选择性TBK1抑制剂进入临床研究,多数TBK1抑制剂研究集中于对2-氨基嘧啶类结构骨架的结构优化。发现新型高选择性TBK1抑制剂仍十分必要,一方面,能够为进一步探究TBK1介导的生物机制研究提供重要的小分子探针,验证其作为肿瘤免疫治疗靶标的作用机制和可靠性;另一方面,也为针对TBK1的肿瘤免疫治疗提供药物候选化合物。
