二硫化钼自支撑电极电化学析氢性能研究
2019-09-16周丹刘灵惠王爱琴银建中
周丹,刘灵惠,王爱琴,银建中
1. 大连理工大学 化工机械与安全学院,辽宁 大连 116024
2. 中国科学院 大连化学物理研究所,辽宁 大连 116024
近年来,电解水制氢引起了人们的极大关注。水分子中氢的质量分数达11%,且其分解后只产生H2和O2,相比于光催化及光电催化,电催化分解水具有较高的制氢效率。与化石基的制氢工艺相比,电解水制氢工艺没有地域限制,可以在任何有电和水的地方使用,并且其氢气的产量和速率可以按需调节。该工艺可以小规模生产,并且能够获得高纯度的氢气(>99.9%),尤其适合小型的加氢站使用,为燃料电池等提供氢源。
提高电化学析氢效率的关键在于电解水的催化剂。目前最有效的电化学析氢催化剂为Pt系贵金属催化剂,一般使用20% Pt/C。然而Pt价格昂贵,地壳丰度低,因此人们尝试减小贵金属粒径,提高活性位暴露等方法来降低贵金属的用量。如单原子催化剂,将Pt的粒径减小到原子级别,从而大大减少了贵金属的用量。大连化物所的包信和课题组[1]制备了单原子的Pt掺杂的MoS2催化剂,并通过理论计算认为单原子Pt的掺杂调变了其周围层内S原子对H的吸附能,从而大大提高了反应活性。Esposito等[2]将单原子层的Pt、Au、Pd等贵金属包裹廉价金属碳化物等,在大大降低贵金属用量的同时获得了与块体贵金属相当的反应活性。另外人们也在积极寻找贵金属的替代品。层状过渡金属二硫化物(layered transitionmetal dichalcogenides,LTMDs),是类石墨烯层状材料,其层间由S-M-S结构组成,并且层与层之间只有弱的范德华力作用,因而可以实现剥离制得单层。2005年,Hinnemann等[3]通过计算发现MoS2的边界位点的ΔGH与Pt相当,随后实验证明MoS2具有很高的析氢催化活性,受到了广泛关注[4]。随后的理论计算和实验都证明了MoS2的活性位点位于其含有不饱和硫原子的边缘处[5]。Huang等[6]通过改变晶格结构、选择性暴露不同晶面来调变MoS2的析氢性能,发现与(Mo2S12)2-团簇有相同结构的晶面结构表现出与Pt相当的氢吸附自由能。
由于二硫化钼电导率较低,电化学析氢活性和稳定性仍然不尽如人意。将二硫化钼制成自支撑电极可以克服这一缺点,如人们尝试将MoS2长在碳布或碳纸上。Xie等[7]将MoS2直接长在碳纸上,并且通过加入DMF,使MoS2纳米片实现了垂直排列,用于Na离子电池的独立电极。由于暴露了更多的S边活性位,这种电极具有更高的可逆电容、初始库伦效率,并且具有优异的循环使用性能。Huang等[8]通过在350 ℃下热解NaH2PO2处理长在碳布上的MoS2,使MoS2上掺杂了P。这种非金属元素的掺入,改变了惰性底面的催化活性,且活化了S边位,其析氢活性得到大大提高。Luo等[9]通过使用Pt电极作对电极,成功地将痕量的Pt电镀到了碳布工作电极上。虽然仅有1.26%的Pt载量,但起始电位为0 mV,Tafel斜率仅为49 mV·dec-1,可以与20%的市售Pt/C催化剂媲美。
本文通过不同的制备方法制备了自支撑MoS2电极,并掺杂镍、钴等非贵金属,改变MoS2的电子特性,从而改善其电化学析氢活性。
1 实验部分
1.1 材料制备方法
1.1.1 四硫代钼酸铵制备方法
圆底烧瓶中称取7.5 g仲钼酸铵,加入100 mL 20%硫化铵水溶液,搅拌至完全溶解(在通风橱进行)。静置2 h,得到暗红色立方晶体及发黑针状晶体。抽滤、用乙醇清洗数次、真空50 ℃干燥过夜[10]。
1.1.2 MoS2制备方法1
0.309 g(0.25 mmol)仲钼酸铵、0.57 g(7.5 mmol)硫脲、36 mL水搅拌15 min至全溶,转移至100 mL带四氟内衬的水热釜中,放入清洗过的1 cm×2.5 cm碳纸,200 ℃加热24 h,得到的碳纸和粉末分别用水和乙醇清洗3次,60 ℃烘干[11],记为钼酸铵MoS2。F-MoS2是在制备过程中加入0.016 g氟化铵(n(F)/n(F+Mo)=0.2)。
1.1.3 Co、Ni掺杂 MoS2制法
0.309 g(0.25 mmol)仲钼酸铵、0.57 g(7.5 mmol)硫脲、36 mL水搅拌15 min至全溶,分别配制0.570 mg·mL-1Co(NO3)2及 0.116 mg·mL-1Ni(NO3)2水溶液,将其以一定剂量加入到上述仲钼酸铵溶液中,搅至混合均匀,转移至100 mL带四氟内衬的水热釜中,放入清洗过的1 cm×2.5 cm碳纸,200 ℃加热24 h,得到的碳纸和粉末分别用水和乙醇清洗3次,60 ℃烘干。36.9%、0.369%、0.018%及0.0018% 的 Co-MoS2中,加入 Co(NO3)2的质量分别为67、6.7、0.26及 0.026 mg。而 0.367%、0.018%和 0.0018% 的 Ni-MoS2中,加入 Ni(NO3)2的质量分别为5.9、0.254和0.025 4 mg。
1.1.4 MoS2制备方法2
0.029 36 g (NH4)2MoS4、30 mL 水或 DMF,转移至50 mL水热釜中,放入清洗过的1 cm×2.5 cm碳纸,200 ℃保温15 h,得到的碳纸和粉末分别用水和乙醇清洗3次,60 ℃烘干,记为H2O-MoS2或DMF-MoS2。
1.2 电化学测试方法
采用三电极体系,工作电极为制备的碳纸电极,用不锈钢电极夹夹住,然后用封口膜包覆,仅暴露1 cm2的电极面积。对电极为石墨棒电极,参比电极为Ag/AgCl电极。电化学测试在Autolab电化学工作站上进行,电解液为0.5 mol·L-1的硫酸水溶液,测试过程始终通入高纯氮气进行饱和处理,测试温度控制为25 ℃。线性扫描伏安曲线测试时,扫描速率为2 mV/s。进行循环伏安扫描时扫速为20 mV/s。
2 结果与讨论
2.1 仲钼酸铵制备MoS2自支撑电极
通过调控仲钼酸铵的浓度分别为4、8、16 mg/mL,制备了3种不同载量的自支撑电极load1、load2、load3,载量分别是 11.2、5.6、4.8 mg/cm2。从图 1可以看到,载量越高,析氢活性越好,而前驱物浓度则和载量成反比。这是因为当前驱物浓度较高时,成核速度较快,没有实现在碳纸上的附着就已经形成大量晶核,所以溶液中MoS2粉末较多,而实际长在碳纸上的较少。因此在制备时应该尽量选取较低的前驱体浓度。

图1 不同MoS2载量的极化曲线
在制备MoS2时,添加一定量的硝酸钴或硝酸镍,再经过进一步的水热,可以将钴和镍掺杂到MoS2中,改变MoS2的析氢活性,如图2所示。
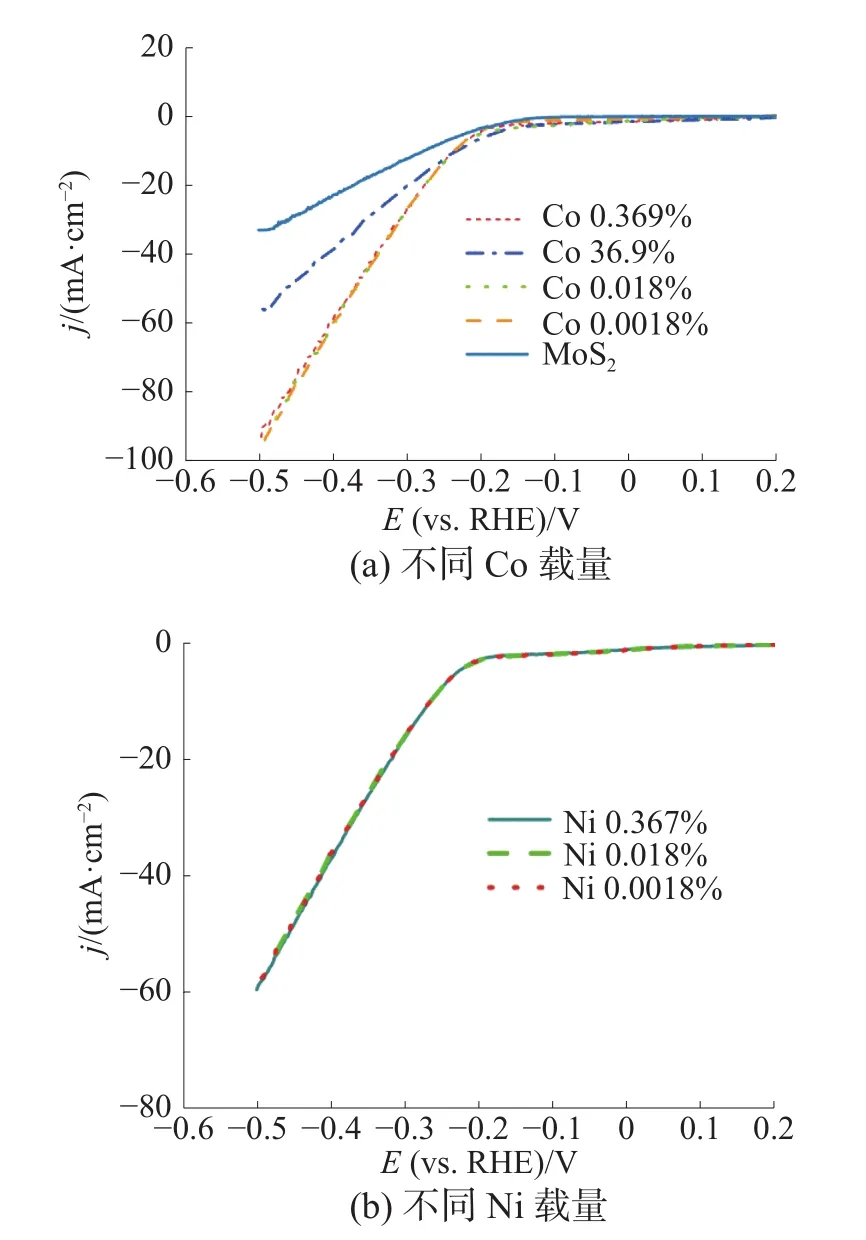
图2 掺杂的MoS2析氢LSV
图2(a)为掺杂一定量的Co的线性伏安曲线。从图可以看到,掺杂量为36.9%时,电流密度为10 mA/cm2时的电位由273 mV降到231 mV;电流密度为20 mA·cm-2时的电位由368 mV降到295 mV。继续降低Co的负载量至0.018%时,电流密度为20 mA·cm-2时电位进一步降低至273 mV;电流密度为10 mA·cm-2时电位仅为234 mV。这是因为掺杂Co较多时,生成的CoS和MoS2同时参与了电化学析氢过程(如图3所示),有锐利边缘的纳米片形状为MoS2,而有明显棱柱状结晶的为CoS。CoS的析氢活性优于MoS2,所以析氢活性整体较高[12]。尽一步降低载量时,Co的电子结构可能影响了MoS2本身的电子结构,改变了电子的传递性能,即提高了电导率,而当载量降低至0.018%时,实现了原子级掺杂。

图3 MoS2的SEM图像
同样方法制得的掺杂Ni的自支撑电极(图2(b)),其电化学析氢活性较MoS2虽然有所改善,但改变载量其电流并没有再发生变化。且同样的载量下,Co掺杂的MoS2表现出优于Ni掺杂的MoS2的活性。
2.2 不同制备方法的比较
采用不同的方法制得了几种MoS2,一种是仲钼酸铵水热法,标记为钼酸铵MoS2;一种是四硫代钼酸铵水热(H2O-MoS2)或将水替换成DMF作溶剂的溶剂热法(DMF-MoS2);还有一种是在仲钼酸铵水热法中加入一定量的氟化铵[13],标记为F-MoS2。图4比较了各种方法制得的MoS2的析氢性能。

图4 不同方法制得MoS2的析氢极化曲线及其Tafel斜率
从图4(a)可以看到,DMF溶剂热制得的MoS2起始电位最低,10 mA电位为220 mV。这是由于DMF作为溶剂时,得到的MoS2均垂直生长于碳纸上,暴露了更多锐利的硫边位[7]。而钼酸铵MoS2、F-MoS2、H2O-MoS2分别为251、281、329 mV。但F-MoS2在-500 mV时的电流密度却远高于DMF-MoS2,它的 Tafel斜率为 62 mV/dec。HER 的基元步骤分为两步:首先,在阴极表面发生电化学反应,质子或水分子接受电子生成吸附的氢原子,即Volmer步骤;随后,吸附的氢原子或通过电化学脱附,即在电子辅助下与另一个质子或水分子发生反应生成氢分子从表面脱附(Heyrovsky步骤),或通过复合脱附,即2个吸附的氢原子复合成氢分子从表面脱附。其中,Volmer步骤为HER必经之路,第二步的脱附路径则由电极催化剂的性质决定。根据速率控制步骤的不同,HER的机理可以分为3种:迟缓放电机理,反应路径为V-T或V-H,其中Volmer为速控步;复合脱附机理,反应路径为V-T,其中Tafel为速控步;电化学脱附机理,反应路径为V-H,其中Heyrovsky为速控步。大量的实验表明,析氢过电位与电流密度之间呈现塔菲尔关系,即:

式中:a和b为实验常数,其中b称为Tafel斜率,可以用来判断HER机理。当b=118.0 mV·dec-1时,电化学反应步骤为决速步,为迟缓放电机理;当b=29.5 mV·dec-1时,为复合脱附机理;当b=39 mV·dec-1时,为电化学脱附机理。从图 4(b)可以得知,H2O-MoS2、F-MoS2以及 DMF-MoS2上发生的析氢过程均属于V-H机理,而F-MoS2的Tafel斜率最低,说明其上发生的电子传输最快。钼酸铵MoS2的Tafel斜率大于118,说明电化学反应为决速步骤。F-MoS2之所以电子传输较快,可能是因为少量的F掺杂到了MoS2的晶格间,配位环境发生了改变。由于F的电负性远大于S,造成Mo电荷偏移,从而加快了电子传输。
3 结论
二硫化钼作为一种有望代替Pt基贵金属的电化学析氢催化剂,其性能改进具有深远的意义。目前主要的改进方法有2种:一种是提高其比表面,使更多的活性位点暴露,常用的方法为锂插层或液相剥离制备单层或少层的MoS2,但锂插层法具有爆炸危险,而液相剥离法制备MoS2产率极低;另一种为通过掺杂和表面修饰,使其电子结构发生改变,促进电子传输。
本文通过溶剂热方法制备4种不同性能的MoS2,并在其上成功地掺杂了非贵金属Ni和Co,改善其析氢性能。
1)过渡金属的掺杂可以明显改善MoS2的析氢活性,而掺杂量仅为0.018%,实现了原子级掺杂;
2)掺杂量相同的情况下(0.018%),Co-MoS2的析氢活性优于Ni-MoS2;
3)DMF-MoS2具有独特的垂直排列形貌,因而暴露出更多的硫边,因此析氢起始电位最低;
4)对F-MoS2来说,由于F的掺入改变了其配位环境,电子传输较快,Tafel斜率最低。
