不同前处理方法对补骨胶囊钙含量测定影响的对比研究
2019-09-12
1.贵州中医药大学,贵州 贵阳 550025;2.贵州广济堂药业有限公司,贵州 贵阳 550014;3.贵州中医药大学第一附属医院,贵州 贵阳 550001;4.贵州省食品药品检验所,贵州 贵阳 550004
伴随着社会的进步,人口老龄化问题愈发严重。骨质疏松的发病率呈逐年上升趋势,并引起了社会的广泛关注[1-2]。为改善人群骨健康状态,本课题组研制开发以增加骨密度、防治骨质疏松为主要功能的保健食品补骨胶囊。处方中中药骨碎补、杜仲、鹿骨粉已被国家卫生健康委员会列为可用于保健食品的中药名单,再与现代常用增加骨密度的营养补充剂骨胶原蛋白粉、碳酸钙、维生素D3相结合,增强产品防治骨质疏松的功能。钙作为维持骨骼韧性和提供骨骼营养的重要物质,是人体必需的成分,对处方增加骨密度,防止骨质疏松的功能有一定的辅助作用,同时,保健食品中钙的含量也是影响产品质量的重要指标[3]。因此根据国家食品药品监督管理局颁布的《保健食品注册与备案管理办法》相关规定,对处方中功效成分钙进行含量测定,以保证产品质量可控。
参考《食品安全国家标准食品中钙的测定》[4]中火焰原子吸收法光谱法建立补骨胶囊中钙含量测定方法。现行钙含量测定标准中样品前处理方法包含微波消解、湿法消解、干法灰法等方法[5-9],在处理过程中,标准中对于未消解完全的样品采用再次消解进行处理,但对于难于消解的,如胶类等,采用多次消解,消解不完全,消解液有颜色、沉淀,对检测结果干扰较大,检测时易堵塞进样系统,对仪器影响大。本试验针对此类问题,进行了摸索,在样品消解过程中,加入适量的30%过氧化氢,使其消解后样品颜色澄清、无沉淀,减少杂质干扰,样品完全消解。本文通过火焰原子吸收法光谱法比较微波消解、湿法消解、干法灰法三种样品前处理方法对补骨胶囊样品消解完全进行对比分析研究,从而建立补骨胶囊最佳钙的含量测定样品前处理方法,为该制剂的质量稳定、可控提供有力依据,并为相关保健食品中钙的含量测定方法提升提供研究依据。
1 仪器与材料
1.1 仪器 AA800原子吸收光谱仪(美国PE公司)、MARS-X微波消解仪(美国玛氏公司)、SX-4-10型箱式电阻炉控制箱(天津市泰斯特仪器有限公司)、BHW-09C可调式电炉(上海博通化学科技有限公司)、ABZ04-S型电子天平(上海梅特勒有限公司)。
1.2 试药 补骨胶囊(批号:20180510、2018-0511、20180512)、碳酸钙对照品(上海麦克林生化科技有限公司,纯度为99.99%,批号:20140424)、氧化镧(上海跃龙化工厂,分析纯,批号:Q/12HB 3875-2009)、硝酸(上海沃凯生物技术有限公司,优级纯,批号:20170623)、盐酸(国药集团化学试剂有限公司,优级纯,批号:20100428)、高氯酸(国药集团化学试剂有限公司,优级纯,批号:20120626)、30%过氧化氢(上海沃凯生物技术有限公司,优级纯,批号:20170623)、水为实验室自制超纯水。
2 方法与结果
2.1 测定方法 参考《食品安全国家标准食品中钙的测定》,采用火焰原子吸收光谱法测定本品钙的含量。
2.2 对照品溶液的制备 精密称取碳酸钙2.4963 g,加适量盐酸溶液(1+1)溶解,置干燥的1000 mL容量瓶中,用水定容至刻度,摇匀,即得钙标准储备溶液(C=1000 mg/L)。准确吸取钙标准储备溶液10 mL置干燥的100 mL容量瓶中,用硝酸溶液(5+95)定容至刻度,摇匀,即得钙标准中间溶液(C=100 mg/L)。
2.3 供试品的制备
2.3.1 微波消解 取补骨胶囊内容物研细,精密称取0.2 g于微波消解罐中,加入5 mL硝酸,浸泡过夜。置微波消解仪进行消解(消解参考条件:120 ℃升温5 min,恒温15 min;160 ℃升温5 min,恒温20 min;190 ℃升温5 min,恒温35 min),冷却后取出消解罐,在可调式电炉赶酸(赶酸参考条件:140 ℃/1h、升至150 ℃/1h、升至160 ℃),消化液赶酸至1 mL左右,颜色呈无色透明,若消化液颜色较深,再加5 mL硝酸,1 mL 30%过氧化氢继续消解、赶酸。赶酸结束后,取出消解罐,放冷,用水润洗消解罐3次,合并洗液至25 mL容量瓶中,用水定容至刻度,即得供试品消解液。根据所需浓度,精密量取供试品消解液0.05 mL,置干燥的50 mL容量瓶中,同时加入2.5 mL 20 g/L镧溶液,混合均匀,用水定容至刻度,使镧在其稀释液中的浓度为1 g/L,即得供试品稀释液,待用。同法做试剂空白溶液。
2.3.2 湿法消解 取补骨胶囊内容物研细,精密称取0.2 g于消化管中,加入10 mL硝酸及0.5 mL高氯酸,浸泡过夜。在可调式电炉消解(消解参考条件:120 ℃/1h、升至180 ℃/3h、升至210 ℃),消化液消解至1 mL左右,颜色呈无色透明或略带黄色,若消化液颜色较深,在加10 mL硝酸继续消解。消解结束后,取出消化管,放冷,用水润洗消化管3次,合并洗液至25 mL容量瓶中,用水定容至刻度,即得供试品消化液。根据所需浓度,精密量取供试品消化液0.05 mL,置干燥的50 mL容量瓶中,同时加入2.5 mL 20 g/L镧溶液,混合均匀,用水定容至刻度,使镧在其稀释液中的浓度为1 g/L,即得供试品稀释液,待用。同法做试剂空白溶液。
2.3.3 干法灰化 取补骨胶囊内容物研细,精密称取0.2 g置坩埚中,小火加热,炭化至无烟,转移至马弗炉中,与550 ℃灰化4 h,样品呈白灰状,冷却,取出,用适量硝酸溶液(1+1)溶解转移至25 mL容量瓶中,用水润洗坩埚3次,合并洗液至容量瓶中,用水定容至刻度,即得供试品消化液。根据所需浓度,精密量取供试品消化液0.05 mL,用水定容至50 mL容量瓶中,同时加入2.5 mL 20g/L镧溶液,混合均匀,用水定容至刻度,使镧在其最终稀释液中的浓度为1 g/L,即得供试品稀释液,待用。同法做试剂空白溶液。
2.4 仪器条件 波长:422.7 nm;狭缝:0.7 nm;灯电流:10 mA;燃烧头高度:3 mm;空气流量:10 L/min;乙炔流量:2 L/min。
2.5 方法学考察
2.5.1 线性关系考察 分别吸取“2.2”项下钙标准中间溶液0 mL、0.5 mL、1 mL、2 mL、4 mL、6 mL于100 mL容量瓶中,同时在各容量瓶中加入5 mL 20 g/L镧溶液,分别加(5+95)硝酸溶液定容,混匀。配制钙标准系列溶液中钙的质量浓度分别为0 mg/L、0.500 mg/L、1.00 mg/L、2.00 mg/L、4.00 mg/L、6.00 mg/L。分别取适量溶液注入仪器,按“2.4”项下仪器条件测定。以浓度为横坐标,吸光度值为纵坐标,计算回归方程为:y=0.0289x-0.00005,r=1。结果表明,钙在0~6.00 mg/L范围内线性关系良好。钙标准曲线,如图1所示。

2.5.2 精密度试验 取补骨胶囊内容物研细,精密称取0.2 g,分别按“2.3”项下方法制备供试品溶液,按“2.4”项下仪器条件测定,分别连续进样6次,计算样品中钙的含量及RSD。结果表明,微波消解、湿法消解、干法灰化测定钙的含量分别为:258.17 mg/g、273.81 mg/g、261.45 mg/g,RSD分别为:0.72%、0.15%、0.76%,表明仪器精密度良好。
2.5.3 重复性试验 取补骨胶囊内容物研细,精密称取0.2 g,各六份,分别按“2.3”项下方法制备供试品溶液,按“2.4”项下仪器条件测定,计算样品中钙的含量及RSD。结果表明,微波消解、湿法消解、干法灰化测定钙的含量分别为:273.91 mg/g、278.78 mg/g、276.47 mg/g,RSD分别为:2.02%、4.35%、6.27%。结果表明,微波消解处理样品的方法较湿法消解、干法灰化的重复性好。
2.5.4 加样回收试验 根据上述考察结果,选取微波消解样品处理方法进行加样回收率试验。取已知含量的样品6份,每份精密称定0.1 g,分别精密量取碳酸钙对照品溶液27500.0 mg/L 1 mL,按“2.3.1”项下方法制备供试品溶液,共六份,按“2.4”项下仪器条件测定,记录浓度,计算样品加样回收率及RSD。结果表明,平均加样回收率为99.0 %,在回收率限度95%~102%之间,RSD在2 %以内,该含量测定方法回收率较好,符合测定要求。结果见表1。
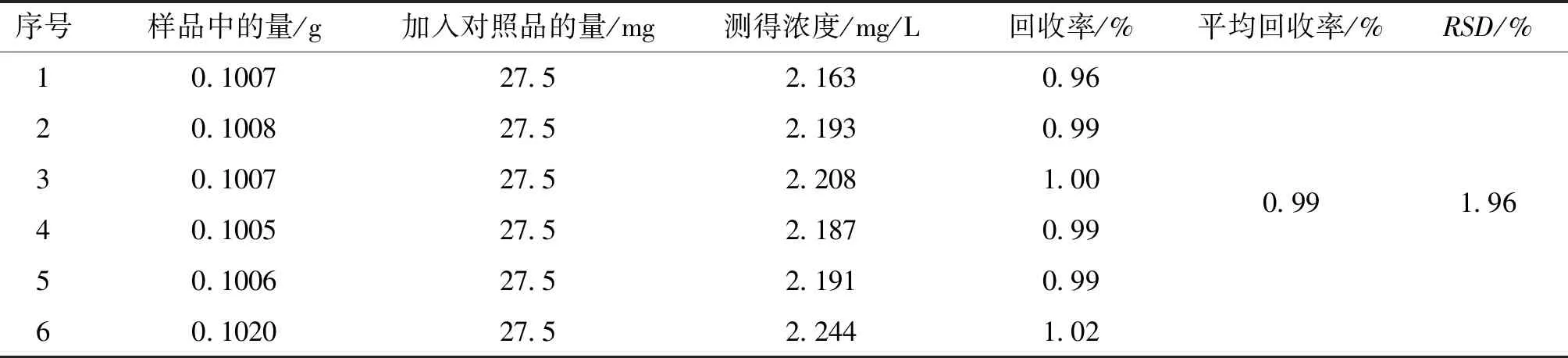
表1 钙回收率试验结果
3 讨论
3.1 钙含量检测方法的比较 现测定钙含量的检测方法主要有EDTA滴定法、火焰原子吸收光谱法、电感耦合等离子体质谱法(ICP-MS)等方法。由于ICP-MS普及度不高,仪器使用条件较高,对样品检测结果产生一定的误差。EDTA滴定法具有快速、简便等优点常作为首选检测方法[10-11]。本试验最先采用EDTA滴定法检测补骨胶囊中钙的含量,试验中发现EDTA滴定法适用于干扰少的样品,但如果样品组成复杂,在滴定时易出现络合物,造成干扰[12-14]。当试验采用火焰原子吸收光谱法检测补骨胶囊中钙的含量较EDTA滴定法结果准确可靠、检测过程干扰小、简单、快速的优点。
3.2 样品消解方法对比及优化 通过火焰原子吸收法光谱法比较微波消解、湿法消解、干法灰法三种样品前处理方法对补骨胶囊样品消解完全进行对比分析研究。试验发现,微波消解、湿法消解、干法灰化三种消解方法中,微波消解比其余两种消解法重复性较好。湿法消解在消化样品时,需加高氯酸强酸强腐蚀强刺激试剂,若前期样品低温消解时有机物消化不完全,遇高氯酸剧烈反应,易喷溅、爆炸,会对操作人员安全造成极大威胁,其样品消化时间长,误差较大,样品检测后钙含量重复性差。干法灰化消解样品时不易消化完全,炭化样品时,容易造成样品污染,样品在测试过程中造成检测数据的波动,其钙含量重复性较差[7]。采用微波消解方法处理样品时,具有速度快,污染少,样品消化完全等特点,结果更加准确可靠[15]。
在试验过程中,因处方中骨胶原蛋白含有一定的胶类物质,不易消解完全,影响钙的含量;消解液不易消解成无色透明状态,影响测定时吸光度变化,故在消解过程中加入适量30%的过氧化氢,可使样品消解完全,减少杂质干扰。
4 结论
综上所述,本试验选择微波消解作为样品前处理方法,方法操作简便,对样品消解完全。应用该方法处理补骨胶囊,进行钙的含量测定,其结果稳定,方法可行,可有效控制补骨胶囊的质量。
