苗药红禾麻野生与引种药材UPLC指纹图谱研究
2019-09-11郭文凯徐文芬孙庆文邹淑涵
郭文凯, 徐文芬, 员 浬, 孙庆文, 王 波, 邹淑涵
(贵州中医药大学, 贵阳 550025)
红禾麻为《贵州省中药材、民族药材质量标准》(2003年版)收载品,贵州少数民族常用药,苗药名为“Uab det dend 蛙斗”等,其来源于荨麻科(Urticaceae)艾麻属(Laportea)植物珠芽艾麻(Lapotreabulbifera(Sieb. et Zucc.)Wedd.)的新鲜或干燥全草,又名红活麻等。具有祛风除湿,活血化瘀等作用,主要用于治疗风湿麻木、跌打损伤、骨折、脾虚、消化不良等症[1]。文献报道,在化学成分方面,汪石丽等[8]、朱珠等[9]共分离鉴定出16个黄酮类和香豆素类化合物。在药理学方面,研究发现红禾麻的提取物总香豆素具有较好的镇痛抗炎和免疫抑制作用[11-16]。在贵州民间常用红禾麻的块根和珠芽泡酒内服和外搽治疗风湿关节痛、坐骨神经痛、骨折等,其成方制剂“润燥止痒胶囊”在治疗老年血虚引起的皮肤瘙痒方面具有独特疗效,近几年的年产值逐渐增加。课题组前期已对红禾麻资源调查、种质形态变异、与易混品的鉴别、药材品质与环境因子和遗传多样性的相关性分析等方面进行研究[2-7],取得了一定的研究进展。但迄今为止,红禾麻的原材料供给主要是依靠采挖野生资源,随着市场需求量逐年增加,野生资源逐年锐减,药材质量良莠不齐,已不能满足生产需要,为了红禾麻资源的保护和相关产业的可持续发展,课题组正在协同相关企业进行红禾麻优良品种选育及规范化种植研究。鉴于此,通过建立红禾麻药材UPLC指纹图谱测定方法,测定分析10批不同产地红禾麻野生药材、与引种于贵州中医药大学种质资源圃药材之间的指纹图谱差异,以期从整体角度为红禾麻优良品种选育与规范化种植提供研究基础,并为该药材整体质量评价及保证市场用药安全有效提供参考。
1 仪器和试药
1.1 仪 器
Thermo UltiMate-3000型高效液相色谱仪;色谱柱Agilent Eclipse plus C18(3.0 mm×100 mm,1.8μm);AG 135型电子天平(瑞士Mettler-Toledo公司);KQ-500 DE型数控超声波清洗器(昆山市超声仪器有限公司);TG 16-WS高速离心机(长沙平凡仪器仪表有限公司);QE-200克高速万能粉碎机(浙江屹立工贸有限公司);中药色谱指纹图谱相似度评价系统2004 A版、2004 B版(国家药典委员会,以下简称指纹图谱软件)。SPSS 22.0数据统计分析软件。
1.2 试 药
1.2.1试 剂
乙腈(色谱纯,德国默克公司)、甲醇(色谱纯,德国默克公司)、甲酸(色谱纯,CNW公司,批号:J 259 K 114);乙醇、甲醇、乙酸乙酯、石油醚等均为分析纯;实验用水为重蒸馏水。
1.2.2样品来源
供试样品均由本课题组前往各分布地区采集,并经贵阳中医学院孙庆文教授鉴定为荨麻科(Urticaceae)艾麻属(Laportea)植物珠芽艾麻(Lapotreabulbifera(Sieb. et Zucc.)Wedd.)的地上部分。所有样品经干燥后粉碎,存放于干燥器内,备用。其中1~10号为野外采集药材样品,Y 1~Y 10为对应的引种于贵州中医药大学种质资源圃1~2年采集药材样品,详细信息见表1。
表1 红禾麻样品来源信息

样品编号采集时间/(年-月-日)采集地点12013-06-01贵州省赫章县22013-06-01贵州省威宁县32013-07-13贵州省贵阳市42013-08-20贵州省梵净山52013-08-22贵州省松桃县62013-10-03贵州省台江县72013-10-04贵州省剑河县82013-11-17贵州省开阳县92014-06-27贵州省修文县102014-10-23云南省富民县Y12015-07-13贵州中医药大学种源圃Y22015-07-13贵州中医药大学种源圃Y32015-07-13贵州中医药大学种源圃Y42015-07-13贵州中医药大学种源圃Y52015-07-13贵州中医药大学种源圃Y62015-07-13贵州中医药大学种源圃Y72015-07-13贵州中医药大学种源圃Y82015-07-13贵州中医药大学种源圃Y92015-07-13贵州中医药大学种源圃Y102015-07-13贵州中医药大学种源圃
2 方法与结果
2.1 供试品溶液的制备[2]
取药材粉末约1.5 g,精密称定,置于50 mL具塞锥形瓶中,加入70%乙醇溶液15 mL,摇匀,称定重量,超声处理(功率100 W、频率40 Hz)45 min,取出放冷至室温,加70%乙醇溶液补足减失的重量,过滤,滤液置离心机(12 000 r·min-1)离心15 min,取上清液过0.22μm微孔滤膜,即得。
2.2 色谱条件
色谱柱:Agilent Eclipse plus C18(3.0×100 mm,1.8μm);流动相:甲醇(A)-乙腈(B)-0.1%甲酸水(C)溶液,流速为0.3 mL·min-1,梯度洗脱按表2程序进行;柱温为30 ℃;进样量为3μL;检测波长为254 nm。

图1 10批野生红禾麻药材的原始指纹图谱叠加图
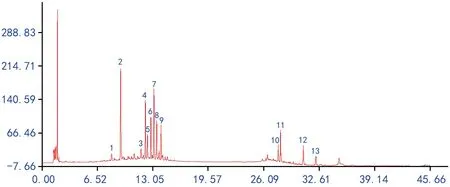
图2 野生红禾麻药材对照指纹图谱
图3 10批引种红禾麻药材的原始指纹图谱叠加图
表2 流动相梯度洗脱系统

T/minA/%B/%C/%03.51.5951055.05.0402155.05.04023100.00.0045100.00.00
2.3 方法学考察
2.3.1空白试验
将提取的70%乙醇溶剂过0.22μm微孔滤膜,进样3μL,按“2.2”项下色谱条件测定。结果表明,采集的色谱图中仅在1.5 min与32.8 min保留时间处有较小的溶剂峰出现,表明提取溶剂及流动相基本无干扰。
2.3.2完整性试验
取药材样品(2贵州省威宁县)约1.5 g,精密称定,依照“2.1”项下制备供试品溶液,按“2.2”项下色谱条件测定,记录45 min后,以100%甲醇流动相继续洗脱至90 min,结果为色谱图中45 min后基本无色谱峰信息,表明采集45 min色谱图即可完整记录红禾麻药材的化学成分信息。
2.3.3精密度试验
取药材样品(2贵州省威宁县)约1.5 g,精密称定,依照“2.1”项下制备供试品溶液,后按“2.2”项下色谱条件,连续进样6次,将所得图谱导入指纹图谱软件A版进行分析,相似度计算结果均大于0.9,表明仪器精密度良好。
2.3.4重复性试验
取同一样品(2贵州省威宁县)6份,每份约1.5 g,精密称定,依照“2.1”项下制备供试品溶液,后按“2.2”项下色谱条件平行测定,将所得图谱导入指纹图谱软件A版进行分析,其相似度计算结果均大于0.9,表明该方法测定重复性好。
2.3.5稳定性试验
取同一供试品溶液(2贵州省威宁县)约1.5 g,精密称定,依照“2.1”项下制备供试品溶液,按“2.2”项下色谱条,分别在0,2,4,6,8,12,24 h依法测定,将所得图谱导入指纹图谱软件A版进行分析,相似度计算结果均大于0.9,表明供试品溶液在24 h内测定稳定性良好。
2.4 样品测定及指纹图谱的建立
在上述的测定条件下,分别取10批不同产地野生红禾麻药材与其各自引种样品粉末,依照“2.1”项下制备供试品溶液,按“2.2”项下色谱条件测定,记录色谱图,分别将10批不同产地红禾麻药材与其各自引种样品的原始指纹图谱的数据文件导入指纹图谱软件A版进行分析,设定时间窗为1.5,采用中位数法,以各自的2号样品图谱为参照图谱,经多点校正后mark峰自动匹配后分别生成不同产地红禾麻药材与各自引种样品的对照指纹图谱,其中不同产地红禾麻药材标定13个共有峰,引种品药材标定10个共有峰,其中不同产地红禾麻与引种品的2号共有峰峰形与分离度较好,故确定2号共有峰为参照峰,结果见图1~图4。
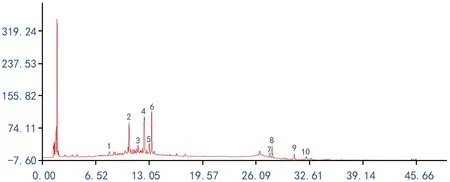
图4 引种红禾麻药材对照指纹图谱
表3 10批不同产地野生红禾麻药材 UPLC 指纹图谱相似度计算结果

批次1234567891011.000 0.223 0.488 0.473 0.444 0.297 0.235 0.613 0.268 0.645 20.223 1.000 0.235 0.630 0.350 0.293 0.212 0.297 0.274 0.270 30.488 0.235 1.000 0.557 0.474 0.329 0.212 0.641 0.289 0.636 40.473 0.630 0.557 1.000 0.682 0.331 0.255 0.608 0.328 0.623 50.444 0.350 0.474 0.682 1.000 0.517 0.344 0.557 0.549 0.571 60.297 0.293 0.329 0.331 0.517 1.000 0.297 0.379 0.405 0.313 70.235 0.212 0.212 0.255 0.344 0.297 1.000 0.323 0.438 0.293 80.613 0.297 0.641 0.608 0.557 0.379 0.323 1.000 0.264 0.769 90.268 0.274 0.289 0.328 0.549 0.405 0.438 0.264 1.000 0.299 100.645 0.270 0.636 0.623 0.571 0.313 0.293 0.769 0.299 1.000
表4 10批对应引种红禾麻药材UPLC 指纹图谱相似度计算结果
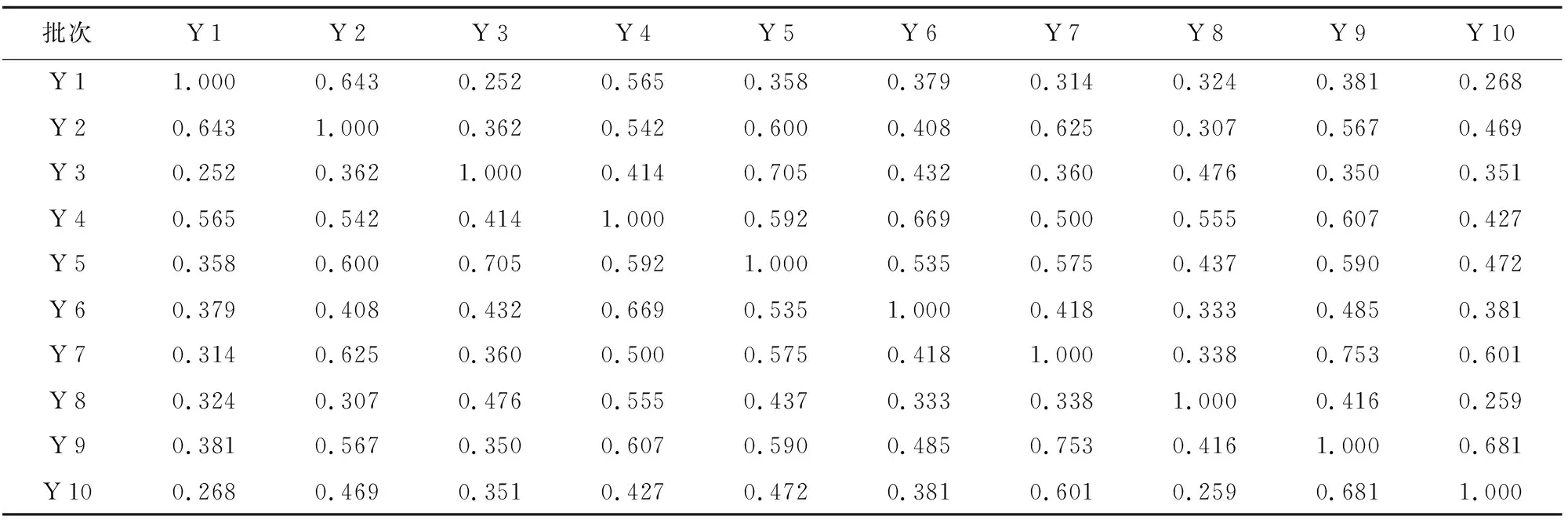
批次Y1Y2Y3Y4Y5Y6Y7Y8Y9Y10Y11.000 0.643 0.252 0.565 0.358 0.379 0.314 0.324 0.381 0.268 Y20.643 1.000 0.362 0.542 0.600 0.408 0.625 0.307 0.567 0.469 Y30.252 0.362 1.000 0.414 0.705 0.432 0.360 0.476 0.350 0.351 Y40.565 0.542 0.414 1.000 0.592 0.669 0.500 0.555 0.607 0.427 Y50.358 0.600 0.705 0.592 1.000 0.535 0.575 0.437 0.590 0.472 Y60.379 0.408 0.432 0.669 0.535 1.000 0.418 0.333 0.485 0.381 Y70.314 0.625 0.360 0.500 0.575 0.418 1.000 0.338 0.753 0.601 Y80.324 0.307 0.476 0.555 0.437 0.333 0.338 1.000 0.416 0.259 Y90.381 0.567 0.350 0.607 0.590 0.485 0.753 0.416 1.000 0.681 Y100.268 0.469 0.351 0.427 0.472 0.381 0.601 0.259 0.681 1.000
2.5 相似度评价
2.5.1红禾麻药材UPLC指纹图谱相似度计算结果
将所测得的原始图谱导入指纹图谱软件A版进行数据分析处理。相似度计算结果表明,10批不同产地红禾麻药材的相似度在0.212~0.769之间,而10批引种品的相似度在0.252~0.753之间,可见引种后的红禾麻药材的内在化学物质变化不大,其相似度计算结果见表3、表4。
2.5.2野生与其对应引种红禾麻药材的UPLC指纹图谱的相似度比较
将所测得的原始图谱导入指纹图谱软件进行数据分析处理,以其野生产地的指纹图谱为参照,进行不同产地野生与其对应引种红禾麻药材的UPLC指纹图谱的相似度比较,由图2和图4可知,引种的对照指纹图谱保留了不同野生品种的1、4、5、7、10、11、12、13号主要色谱峰,因而10批野生与其对应引种红禾麻药材的UPLC指纹图谱相似度大部分在0.75以上,对比其图谱后发现,引种后的环境改变对红禾麻药材化学成分的影响不明显,其中相似度较低的Y 18→8号样品,对比图谱发现,原产地1号峰峰面积为0.370,而引种品为0.995,原产地4号峰为0.135,引种品为0.916,原产地5号峰为3.691,引种品为1.755,原产地7号峰为0.044,引种品为0.645,原产地10号峰为1.660,引种品为0.304,原产地11号峰为2.586,引种品为0.368,因其主要色谱峰峰面积变化较大,故相似度最差,Y 9→9与Y 10→10为同样原因导致,其相似度计算结果见表5。

图5 10批不同产地野生红禾麻指纹图谱聚类分析图
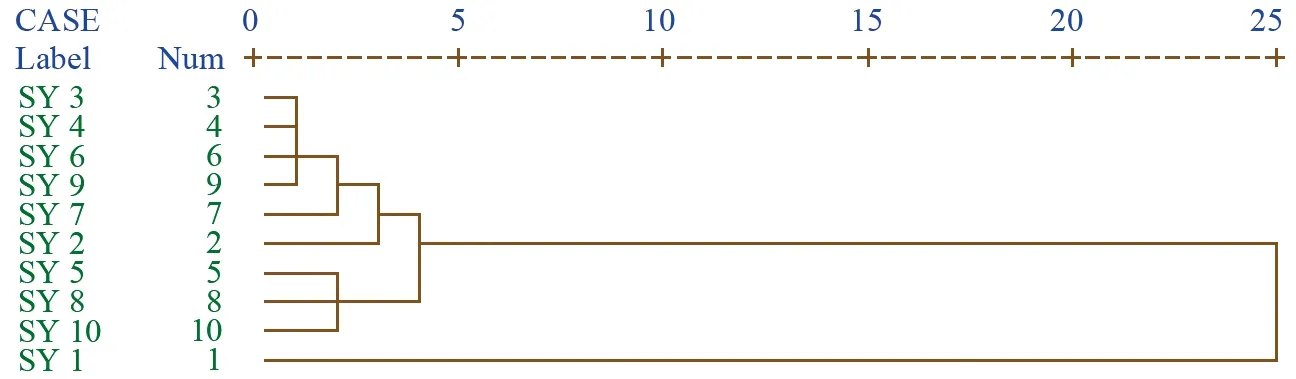
图6 10批引种红禾麻指纹图谱聚类分析图
2.6 聚类分析
将10批野生及其对应引种红禾麻药材指纹图谱中,各色谱峰面积相对于2号参照峰量化,进行系统聚类,由聚类谱系图可以明显看出:在不同产地野生品种中,在欧式距离为5时,3、6、7、8聚为一类,5、9聚为一类,10、2、4、1分别聚类一类,共聚为6类,而在引种品中,在距离为5之前,除Y 1号外,其余的引种品相似度很高,已经归为一类,这充分说明野生红禾麻药材由于产地差异而在药材质量上存在差异,而引种于同一环境下的红禾麻药材则内在质量差异较小。结果见图5、图6。
对比野生红禾麻与引种后的对照图谱可以发现,引种后其主要峰群均较好的存在,而相似度结果表明,引种后的红禾麻与野生的相似度大部分在0.75以上,聚类结果表明引种后的红禾麻在欧式距离为5之前,除Y 1号外已经聚为一类。综合上述分析,建议可以在模拟野生红禾麻生长环境的基础上进行引种,为市场提供质量均一的红禾麻药材。
表5 野生与其对应引种红禾麻药材的UPLC指纹图谱的相似度比较结果

样品相似度Y1→10.926→1Y2→20.870→1Y3→30.826→1Y4→40.773→1Y5→50.829→1Y6→60.811→1Y7→70.807→1Y8→80.313→1Y9→90.523→1Y10→100.591→1
3 讨 论
本试验采用二极管阵列检测器分别比较了254 nm,270 nm,320 nm,360 nm 不同检测波长下的色谱分离情况,结果显示254 nm波长下检测到的色谱峰容量大、信号强、峰形好,且各色谱峰之间分离度较好,故选择254 nm作为检测波长;比较了甲醇-水、乙腈-水、甲醇-乙腈-水、甲醇-乙腈-0.1%甲酸水溶液4个流动相系统进行梯度洗脱,结果表明:以甲醇-乙腈-0.1%甲酸水溶液进行梯度洗脱时,色谱峰个数多,分离度好,峰形也较好。故选择以甲醇-乙腈-0.1%甲酸水溶液为流动相;分别考察了25 ℃、30 ℃、35 ℃的柱温,结果显示:柱温为30 ℃时,色谱峰分离度较好,响应值较高且峰形较好。故选择30 ℃作为检测柱温;分别设置进样量为3μL、4μL、5μL,结果显示:进样量为3μL时,色谱峰分离度较好,且各进样量之间的色谱峰个数无明显区别,故选择3μL的进样量。故最后确定色谱条件为色谱柱:Agilent Eclipse plus C18(3.0 mm×100 mm,1.8μm);流动相:甲醇(A)-乙腈(B)-0.1%甲酸水(C)溶液,流速为0.3 mL·min-1,柱温为30 ℃,进样量为3μL,检测波长为254 nm。
相似度计算结果说明,不同地区的环境对红禾麻的生长有较大影响,造成不同产地的红禾麻药材的内在质量具有一定的差异。而引种品与原产地红禾麻药材的相似度大部分在0.75以上,初步说明引种后的生长条件与红禾麻原产地相似,适宜红禾麻药材的生长,这可以为红禾麻药材的引种提供参考依据。
聚类分析结果说明,由于3、6、7、8号红禾麻分别来源于贵阳、台江、剑河、开阳,地理距离相隔比较近且10号共有峰的峰面积为1.5左右故聚为一类,而10、2、4、1各聚为一类,则是由于它们在地理位置上相隔较远、海拔相差较大、共有峰面积变化较大而单独分类。而在引种品中,在距离为5之前,除Y 1号外,已经归为一类,这可能由于引种后的红禾麻在同一环境中生长,药材趋于同质,更加适宜红禾麻药材在源头进行质量控制。
本试验所建立的红禾麻药材的超高效色谱指纹图谱具有一定的专属性和重现性,并且较高效液相色谱节约试剂与时间,可以初步作为红禾麻药材的化学鉴别依据和质量控制依据,可为红禾麻药材的野生驯化、优良品种选育与规范化种植研究提供指导依据。
